
Translate this page into:
A rare alpha chain variant hemoglobin St Luke’s reported for the first time in India – A case report
*Corresponding author: Kainaz Sidhwa, Department of Hematology, Bhide Laboratory Services, Mumbai, Maharashtra, India. drkainazsidhwa@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Shinde P, Sidhwa K, Nadkarni A, Chiddarwar A. A rare alpha chain variant hemoglobin St Luke’s reported for the first time in India – A case report. J Hematol Allied Sci. 2025;5:93-5. doi: 10.25259/JHAS_10_2024
Abstract
The ever-increasing gamut of hemoglobin (Hb) variants comprises a majority that appear to be both clinically and hematologically silent, but often present with difficulties in interpretation of high-performance liquid chromatography histograms as well as Hb electrophoresis. The challenges faced in the reporting and characterization of these Hbopathies are circumvented by the use of molecular techniques. Here, we report a rare alpha chain variant, Hb St Luke’s that was incidentally detected during routine laboratory work-up carried out for an adult male patient. We report the first case of this Hb variant from India.
Keywords
Alpha chain variant
Hemoglobin St Luke’s
Rare hemoglobin

INTRODUCTION
Hemoglobinopathies are the most common single-gene disorders to affect population all over the world. To date, there are more than 1700 hemoglobin (Hb) variants that have been reported, with more than 900 variants of the β-globin gene alone.[1] Structural Hb variants causing qualitative defects are invariably caused by single amino acid substitutions. In other cases, two amino acid substitutions in the same globin chain, N or C terminal elongation, deletions, insertions, or hybrid globins are the cause of the resultant variant Hb.[2] While arguably most of them are clinically and hematologically silent, their correct identification and prognostication are detrimental, especially when there are variants that can result in a frank hemolytic anemia or thalassemia phenotype especially when found in association with other hemoglobinopathies. The most common by far being the sickle syndromes. It is well established that many non-HbS Hbs that can be either alpha or beta-globin variants, elute at similar retention times as that of the HbS window (4.3–4.7 min) in high-performance liquid chromatography (HPLC).[3,4] This diagnostic conundrum faced by many often results in the discovery of rare hemoglobinopathies as seen in our case. We report for the 1st time Hb St Luke’s detected in an adult Indian male.
Hb St. Luke’s (α95[G2]Pro→Arg) (A1) or HBA1: c.287C>G, is an α-globin chain variant of the α1 globin gene. It is a rare Hb variant that has been described originally in Maltese and later in Japanese patients.[5-7] Hb St. Luke’s results from a substitution of Arginine for Proline at codon 95 in the G helix of α1-globin chain results in the Hb molecule with less negative charge as compared to the Hb A. In a study done by Molchanova et al. on differences in quantities of α2- and α1 globin gene variants in heterozygotes, Hb St. Luke’s was characterized (Mildy) as an unstable variant.[8]
CASE REPORT
A 56-year-old Gujrati Jain male who was incidentally detected to have an abnormal variant during assessment of HbA1c underwent HPLC to further investigate and analyze the Hb variant. HPLC findings revealed an abnormal Hb (10.8%) eluting in the “S” window with a retention time of 4.49 min [Figure 1].
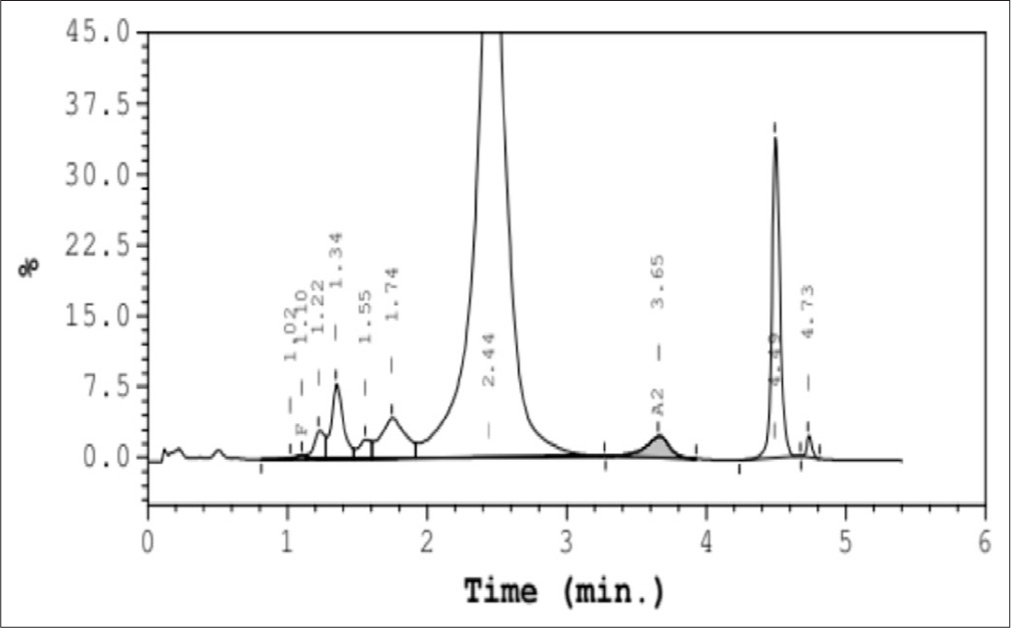
- High performance liquid chromatography of hemoglobin St. Luke eluting in the “S” window.
The sickling test was found to be negative suggesting the possibility of an unknown Hb. Evaluation of the low percentage and retention time of this unknown Hb variant detected with a negative sickling test led to the possibility of an unknown alpha chain variant.
For confirmation of diagnosis, the sample was subjected to direct DNA sequences of the amplified α-globin gene in four fragments. We reported substitution CCC➜CGC at codon position 9 which resulted in substitution of amino acid Proline by Arginine. This mutation led to the production of abnormal Hb Hb St. Luke’s [Figure 2].
![Deoxyribonucleic acid sequencing of α-globin gene showing hemoglobin St. Luke’s [α95[G2]Pro→Arg]. A: Adenine, G: Guanine, C: Cytosine, T: Thymine, Pro: Proline , Arg : Arginine.](/content/129/2025/5/1/img/JHAS-5-093-g002.png)
- Deoxyribonucleic acid sequencing of α-globin gene showing hemoglobin St. Luke’s [α95[G2]Pro→Arg]. A: Adenine, G: Guanine, C: Cytosine, T: Thymine, Pro: Proline , Arg : Arginine.
DISCUSSION
Hb-St Luke’s was detected for the 1st time by routine starch-gel electrophoresis in four members of a Maltese family. The carriers of this abnormality were clinically and hematologically normal.[5]
A couple of years later Lorkin et al. reported a case of Hb St. Luke’s once again detected in a Maltese man. He also had polycythemia.[9] As polycythemia is sometimes associated with a high-affinity Hb, the authors have examined the oxygen affinity of isolated Hb St. Luke’s in phosphate buffer and have found it to be increased.
In 1981, a study was conducted to assess the production of five alpha chain variants in heterozygotes among members of the same family. The 12 Hb St. Luke’s heterozygotes who participated in the study were Caucasians from Malta.[10]
Harano et al. reported the third case of Hb St. Luke’s. This was the first case of Hb St Luke’s encountered among the Japanese people.[7] Analysis of the variant Hb revealed high oxygen affinity and a tendency toward dissociation of tetrameric molecules into dimers due to a weakened α1β2 contact. Their findings were comparable to Lorkin et al [9] as well as Bezzina Wettinger et al.’s study done on the characterization and locus assignment of alpha globin genes.[6] In this study, Hb St Luke’s comprised 11 cases of the 23,000 Maltese neonates and pregnant women screened, while Hb St Luke’s is an α-globin chain variant of the α1 globin gene. Singha et al. reported an un-described α-globin gene triplicated allele with a novel Hb variant on α2-globin gene, Hb St. Luke’s-Thailand [α95(G2)Pro-Arg]. They speculated that although these occurred on different α-globin genes, Hb St. Luke’s and Hb St. Luke’s Thailand have similar hematological manifestation and phenotypic identity. Both of them are likely non-pathological Hb variants.[11]
Although the list of documented and recorded Hb variants is large,[12] it is also expansible with newer variants continually added. Moreover, while all Hb variants do not pose a significant clinical threat, it is still imperative that they are diagnosed with accuracy to further determine the clinical prognosis and possible prevention of deleterious combinations by prenatal testing. Even the clinically insignificant Hb variant holds importance when it mimics a clinically significant Hb’s HPLC findings as seen in our case. Of the novel and rare Hbs reported from India, Hb Titusville, Hb Shimonoseki, Hb Chandigarh, Hb D Agri, Hb Yaizu, Hb Rush, and Hb Vellore are Hbs that elute in the HbS window on HPLC.[13] Of these, only the Hb Titusville and Hb Shimonoseki are alpha chain variants. With our case, a third alpha chain variant that elutes in the HbS window is added to the list.
The importance of correctly diagnosing a Hb variant is to evaluate and prevent co-inheritance with other hemoglobinopathies or hematological disorders. With the reporting of rare Hb variants, their existence and significance come to light, such that one can keep a high index of suspicion if faced with similar diagnostic scenarios.
CONCLUSION
As we progress in our methods of Hb variant detection from electrophoresis and HPLC to molecular methods, the discovery and characterization of newer variants will continue. With future advances in medicine and more sophisticated detection of physiologic derangement, even the stand-alone clinically stable and asymptomatic Hb variant would be of significance.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Research. 2020;49(D1):D1192-6.
- [CrossRef] [PubMed] [Google Scholar]
- Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harbor Perspectives in Medicine [Internet]. 2013;3(3):a011858-8.
- [CrossRef] [PubMed] [Google Scholar]
- HPLC retention time as a diagnostic tool for hemoglobin variants and hemoglobinopathies: A study of 60,000 samples in a clinical diagnostic laboratory. Clinical Chemistry. 2004;50:1736-47.
- [CrossRef] [PubMed] [Google Scholar]
- Detection of abnormal hemoglobin variants by HPLC method: common problems with suggested solutions. Int Sch Res Notices. 2015;2015:498230.
- [CrossRef] [PubMed] [Google Scholar]
- Hemoglobin St Luke's or α95Arg2 (G2) β2. European Journal of Biochemistry. 1972;29(2):301-7.
- [CrossRef] [PubMed] [Google Scholar]
- Characterization and locus assignment of two α-globin variants present in the maltese population: Hb St. Luke's [α95(G2)Pro→Arg] and Hb Setif [α94(G1)Asp→Tyr] Hemoglobin. 1999;23(2):145-57.
- [CrossRef] [PubMed] [Google Scholar]
- Hb St. Luke's [α 95 (G2) Pro replaced by Arg] in Japan. Hemoglobin. 1987;7:471-2.
- [CrossRef] [PubMed] [Google Scholar]
- The differences in quantities of alpha 2-and alpha 1-globin gene variants in heterozygotes. British Journal of Haematology. 1994;88(2):300-6.
- [CrossRef] [PubMed] [Google Scholar]
- The oxygen affinity of haemoglobin St. Luke's. FEBS Letters. 1974;39(1):111-4.
- [CrossRef] [PubMed] [Google Scholar]
- Alpha-thalassemia and the production of different alpha chain variants in heterozygotes. Biochemical Genetics. 1981;19(5-6):487-98.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular and hematological characteristics of a novel form of α-globin gene triplication: The hemoglobin St. Luke's-Thailand [α95(G2) Pro→Arg] or Hb St. Luke's [A2] HBA2. Clinical Biochemistry. 2013;46(7-8):675-80.
- [CrossRef] [PubMed] [Google Scholar]
- ITHANET: Information and database community portal for haemoglobinopathies. Bioinformatics. 2017;12:189-191.
- [CrossRef] [Google Scholar]
- Wide spectrum of novel and rare hemoglobin variants in the multi-ethnic Indian population: A review. International Journal of Laboratory Hematology. 2024;46(3):434-450.
- [CrossRef] [PubMed] [Google Scholar]




