
Translate this page into:
Epidemiological features and clinical profile of patients with thalassemia in Kabul, Afganisthan
*Corresponding author: Khwaja Mir Islam Saeed, AFETP Training, Afghanistan National Public Health Institute, Ministry of Public Health, Kabul, Afghanistan. kmislamsaeed@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Saeed KMI. Epidemiological features and clinical profile of patients with thalassemia in Kabul, Afganisthan. J Hematol Allied Sci. 2025;5:54-60. doi: 10.25259/JHAS_28_2024
Abstract
Objectives:
Thalassemia is the most common genetic disorder globally. In Afghanistan, the epidemiology and clinical characteristics of the disease are not studied officially. This study aims to explore the epidemiological pattern and clinical profile of thalassemia patients registered in the National Blood Bank (NBB), Kabul, Afghanistan.
Material and Methods:
A retrospective medical record review was conducted to analyze registries and conduct phone calls for follow-up patients registered in the NBB during 2019–2020 in Kabul. The confirmed case was any patient tested for fetal hemoglobin and blood film. Data were collected using a form matching the patient’s registers. Epi Info v.7 was used for data management and descriptive measures analysis.
Results:
Totally 411 patients with thalassemia were enrolled in the analysis. Out of all patients, 225 (54.61%) were male, with an overall mean age of 6.89 (±4.06) years. The highest proportion of patients was clustered around age groups of 2–8 years (56%). Pashtuns’ ethnicity had the highest percentage of patients (63%) and Hazara the lowest (1%). Almost the majority of cases under management were thalassemia major (98.8%), and just one case was recorded as intermedia and four cases as minor. According to laboratory tests, 88 (21.4%) were positive for hepatitis C, 2 (0.5%) positive for hepatitis B, and 1 (0.2%) for human immunodeficiency virus. As a whole, 251 (61.67%) had a type of consanguinity marriage. In patients, the blood groups of A+ (27%), B+ (27%), and O+ (25%) were almost equally distributed, while AB+ (10%), O negative (4%), A negative (2%), and AB negative (0.2%) had low proportions. The residency of the majority (87.13%) was Kabul. Totally, 18 (4.36%) had surgical operations, and 11.8% reported children died due to thalassemia.
Conclusion:
Thalassemias are common in Afghanistan, and facility for management is lacking. Consanguineous marriage was more common and probably a contributing factor to disease. The establishment of more centers and a full review of factors are recommended.
Keywords
Afghanistan
Blood group
Epidemiology
Marriage
Thalassemia

INTRODUCTION
Thalassemia is a common gene disorder all over the world, particularly in the tropical and subtropic regions.[1] Thalassemia is an inherited blood disorder in which the affected child is unable to synthesize its own hemoglobin, an important part of red blood cells, so the red blood cells do not function properly, traveling in the bloodstream. Thalassemia is classified based on its hemoglobin parts, usually either “alpha” or “beta,” or based on its severity, which is called trait, carrier, intermedia, or major thalassemia.[2] Thalassemia is mostly dominating in the area known as “the thalassemia belt,” ranging from Northwest Africa and the Mediterranean region to Southeast Asia.[3] Traits for thalassemia are more common in people from Mediterranean countries, such as Greece and Turkey, and in people from Asia, Africa, and the Middle East.[2] Most children with thalassemia are born in low-income countries. Worldwide, transfusion is available for a small fraction of those who need it, and most transfused patients will die from iron overload unless an available and potentially inexpensive oral iron chelator is licensed more widely.[4]
An estimated 320,000 babies are born each year with a clinically significant hemoglobin disorder, and more than 100 million beta-thalassemia carriers with a global frequency of 1.5% are conservatively estimated.[1,5] Southeast Asia accounts for about 50% of the world’s carriers, while Europe and the Americas jointly account for 10–13% of the world carriers.[6] Hence, the disorder is common in the Middle East and West Asia and is probably the most common inherited hemoglobin disorder in India. Beta-thalassemia is reported to be between 3% and 7% in most of North Africa.[7] In western countries, thalassemia affects mostly individuals whose ancestries are traceable to a high prevalence area.[8] All types of thalassemia can be fatal in some cases, particularly when multiple gene mutations affect the production of globin chains. In 2013, almost 25,000 deaths were attributed to thalassemia, which was an improvement on the 36,000 deaths recorded in 1990.[9]
In the eastern Mediterranean region, the common autosomal recessive disorders are alpha-thalassemia, with a carrier rate range between 2% and 50%, and beta-thalassemia, with a carrier rate range between 2% and 7%. Furthermore, the high consanguinity rate of 20–50% in most countries of the region has been highlighted as a main predictor of autosomal recessive genetic disorders and hemoglobinopathies, including thalassemia and sickle-cell disease.[10,11] Studies of disorder in Pakistan, the eastern neighbor of Afghanistan, have shown that 5% of the population have thalassemia minor, and the total number of children with thalassemia major may be over 50,000.[12,13] In addition, in Pakistan, most of the children are chronically under-transfused and iron overloaded. The median age at death is just 10 years.[14] The findings from a study in Iran show that the average thalassemia carrier prevalence rate is about 4%[15], while other literature indicated that the prevalence of the disease in a few areas is between 4% and 8%. For instance, in Isfahan, the frequency rises to about 8%, and in the Fars province, in southern Iran, the gene frequency is high and reaches 8–10%.[16] The average prevalence of beta-thalassemia carriers is 3–4%, which translates to 35–45 million carriers in India. Several ethnic groups have a much higher prevalence (4–17%).[17]
Likewise, a child with a thalassemia major is born only when both parents have thalassemia minor. In a carrier couple, there is a 25% probability in each pregnancy that the child may inherit an abnormality from both parents. Marriage of a carrier with a non-carrier will not result in thalassemia major. Thalassemia minor (carrier) is an asymptomatic disorder, and most people do not even know about their abnormality. Most people with mentioned thalassemia are detected during blood testing for some other reason or when they get married to another carrier and give birth to a child with thalassemia major.[18] In an Indian study, strategies to control thalassemia have been listed as educating health professionals, school and college students, pregnant women, and the population at large establishing prenatal diagnosis facilities in different regions of the country, setting up a greater number of centers for managing existing thalassemia patients, and developing cost-effective facilities for stem cell transplantation across the country.[19]
The number of patients with thalassemia is not clear in Afghanistan. Equipped centers and treatment facilities for patients are not adequate. Most of the patients are treated at the centers run by government and nongovernmental organizations that have limited resources. More than 1500 children are suffering from thalassemia in Nangarhar province.[20] In another study by NATO in which 369 subjects were studied, the prevalence of beta-thalassemia was 3.8% in the country.[21] Moreover, it is projected that the number of cases of thalassemia would be rising in Afghanistan, being part of South Asian countries.[22] In addition, in an article, Iran claims that due to the existence of Afghan and Iraqi refugees in Iran, this massive ethnic/genetic commixture has led to an unexpectedly high number of different mutations of β-thalassemia.[23] Afghanistan.[24] According to the World Health Organization (WHO) estimation, there are 14.4% alpha plus thalassemia carriers and 3% beta-thalassemia carriers among Afghanistan pregnant women.
In 2006, the WHO designated thalassemia as a major public health concern;[25] however, accurate information on the health burden of thalassemia with respect to patient numbers, genotypes, treatment requirements, disease-related complications, and mortality in many countries, including Afghanistan is unclear. As a result, providing advice to governments and policymakers about the current or projected disease burden has been a challenge.[26] Due to the years of war and conflict, the culture of research and studies has poorly been established and/or institutionalized in the country. Including the other sections of health, very little information is available on heredity diseases such as thalassemia. Patients with this inherited disorder are prone to infectious and blood-borne diseases, including human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome and hepatitis. It is said that thalassemia patients are prone to malaria as well. Blood transfusion is the only option for the treatment of major thalassemia, which overloads them with iron. It will lead to damage to organs and other complications. After infectious disease, mortality of children due to thalassemia could be an issue of concern in the country. If under treatment, it will affect the quality of life and life expectancy. If an organized data collection and analysis is conducted, it will specify and characterize the patients, which will inform decision-makers and planners to formulate strategic actions for its prevention and control. The key purpose of this study is to review the medical records and describe the epidemiological pattern and clinical profile of thalassemia cases registered in the National Blood Bank (NBB) at the Ministry of Public Health in Kabul city.
MATERIALS AND METHODS
A retrospective medical record review was conducted to study the epidemiological pattern of patients with thalassemia registered in NBB in Kabul city. All records of the clients which have been registered in this setting were reviewed and analyzed for the year (1398) 2019–2020. This study focused on patients of any age and gender who were affected with thalassemia and registered in this center. The data abstractor was trained to extract the data from register books. Furthermore, based on phone numbers, the team contacted patients to make follow-ups and collect more data on the outcome of the disease. The thalassemia unit is located in NBB, being a six-storied building responsible for providing blood services to all patients in Kabul as well as provinces. The unit in NBB is providing just blood transfusion and also anti-chelating agents while diagnosis, medical treatment, and surgical operations are conducted in Indira Ghandi Children Hospital. Diagnosis of thalassemia is based on clinical findings, complete blood count (CBC), and determination of hemoglobin fetal and its percentage by Alkaline denaturation. The patients whose clinical picture and CBC match the thalassemia and fetal hemoglobin is more than 20% are classified as major and eligible for blood transfusion and referred to NBB at the thalassemia unit.
Altogether, protocol development, data collection, data entry, data analysis, and development of the report were completed within 3 months (January–March 2020). As the thalassemia ward was newly established and almost 400 patients were under management, we included total medical records in our review from April 2019 to February 2020 for almost 1 year. The sampling strategies for the abstraction of data from registers were to approach the medical record office of NBB and temporarily barrow the registers, and obtain the data which are planned by the study team. The variables that were collected are almost bound to the fields of the register books, which are available in the medical record office. These variables were age, gender, date of registration, duration of services being provided, physical address, telephone number, family relationship of parents, blood group of father and mother, ethnicity, number of children, number of affected children, laboratory examination, and severity of disease. So, based on variables, a data collection form was developed to abstract data from medical records. A few competent staff from the Afghanistan National Public Health Institute (ANPHI) were trained in data collection and they used the data abstract form to collect data.
Using the data abstracted from a database was developed in Epi Info version 7 to use for data management. Data double entry was done followed by data cleaning, validation, and data analysis with the same software. Thalassemia patients, including males and females of any age who were registered in NBB in Kabul city, were included in the database, while patients whose records were defective and not eligible for analysis were excluded from the study. There was no incentive or compensation, including monetary or non-monetary in this study to participants or to the NBBs. However, the study provided very valuable information that could be used for the formulation of strategies to improve the status of registries and the prevention and control of thalassemia in the country. The issue of quality was ensured from the design of the study until the dissemination of results. Data abstractors were trained, and the investigator worked with them on how to abstract data from registers to ensure quality data. Monitoring was conducted by the principal investigators during data collection and data entry. The research protocol was submitted to the Institutional Review Board at ANPHI to be assessed technically and ethically. After its approval, the implementation was conducted. Furthermore, official permission was taken from the management team of NBB in Kabul. We tried to ensure the confidentiality of the records of patients through deidentification of the patients for data entry and analysis.
We hypothesized the cousin marriage will increase the possibility of having thalassemic children. In addition, patients affected by thalassemia will be prone to infection, and the test will show positive results for hepatitis C (HBC), hepatitis B virus, or HIV. All activities, including designing the protocol, data entry, data analysis, development of the report, and submission of the manuscript, were done using current expertise in ANPHI and the Afghanistan Field Epidemiology Training Program (AFETP) in the country. Stationery, photocopying, and printing were also used in AFETP facilities. Therefore, using available managerial and technical capacity, no extra budget for this study was required.
RESULTS
A total of 413 records of thalassemia patients for almost 1 year in the registries of NBB, were reviewed. As the two records were incomplete, they were excluded, and 411 were included in the analysis. Out of all registered cases, 225 (54.61%) were male and 187 (45.39%) were female. The overall mean age of the patients was 6.89 (±4.06 standard deviation) years, with a differentiation of 7 (±3.8) years in males and 6.7 (±4.3) years in females. However, this difference was not statistically significant. The highest proportion of patients was clustered around age groups of 2–5 years (27%) and 3–8 years (29%). On average, each family had 4.8 (±2.3) children of whom 1.5 (±0.8) children were affected with thalassemia. The proportion of patients with their ethnicity showed that Pashtuns had the highest percentage of patients (63%) and Hazara had the lowest percentage of records (1%). Table 1 depicts more detailed information.
| Variables | Consanguinity (+) | Consanguinity (−) | Total | |||
|---|---|---|---|---|---|---|
| n | % | n | % | n | % | |
| Sex | ||||||
| Male | 141 | 56 | 83 | 53 | 224 | 55 |
| Female | 110 | 44 | 73 | 47 | 183 | 45 |
| Age groups | ||||||
| <=2 | 38 | 15 | 17 | 11 | 55 | 13 |
| >2–5 | 60 | 24 | 48 | 31 | 108 | 26 |
| >5–8 | 82 | 33 | 38 | 24 | 120 | 29 |
| >8–11 | 35 | 14 | 29 | 19 | 64 | 16 |
| >11–14 | 26 | 10 | 15 | 10 | 41 | 10 |
| >14 | 10 | 4 | 9 | 6 | 19 | 5 |
| Number of children | ||||||
| <2 | 52 | 21 | 24 | 15 | 76 | 18 |
| 2–<4 | 76 | 30 | 45 | 29 | 121 | 29 |
| 4–<6 | 80 | 32 | 41 | 26 | 121 | 29 |
| 6–<8 | 30 | 12 | 35 | 22 | 65 | 16 |
| >8 | 13 | 5 | 11 | 7 | 24 | 6 |
| Residence | ||||||
| Kabul | 237 | 94 | 139 | 0.891 | 376 | 92 |
| Provinces | 14 | 6 | 17 | 0.109 | 31 | 8 |
| Ethnicity | ||||||
| Pashtuns | 164 | 65 | 91 | 58 | 255 | 62 |
| Tajiks | 74 | 29 | 47 | 30 | 121 | 29 |
| Hazara | 4 | 2 | 1 | 1 | 5 | 1 |
| Pashayee | 5 | 2 | 11 | 7 | 16 | 4 |
| Others | 1 | 2 | 6 | 4 | 7 | 2 |
Almost the majority of cases under management were thalassemia major 406 (98.8%), and just one case was recorded as intermedia and four cases as minor. There was no hemoglobin electrophoresis to identify the genetic types of thalassemia. The patients were tested for hepatitis B (HBs), HBC, HIV, and Venereal Disease Research Laboratory (VDRL) (Syphilis) when they approached the thalassemia center. The results of the study showed that 88 (21.4%) were positive for HBC, 2 (0.5%) positive for HBs, 1 (0.2%) for HIV, and no one was positive for VDRL after testing in the NBB.
As a whole, 251 (61.67%) had a type of consanguinity marriage, and the rest were not relatives of each other, and just four did not reveal their relationship. However, the team categorized the responses into being first cousins, double first cousins, second cousins, and other far relatives, and being unrelated in terms of consanguinity. Figure 1 shows the number of percentages of this distribution among patients in the registry.
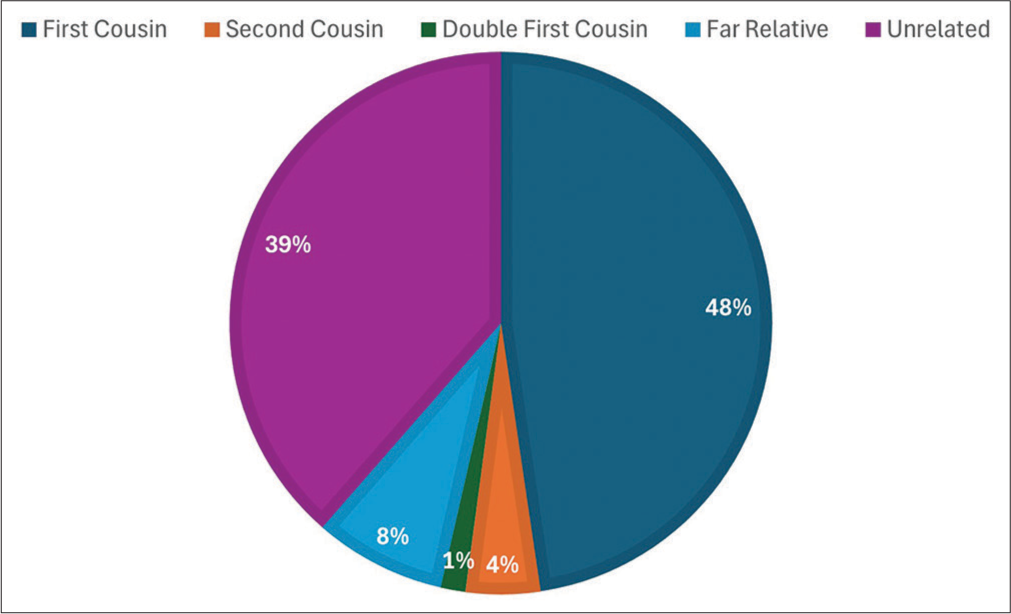
- Proportion of different categories of marriage among patients of thalassemia in Kabul Central Blood Bank, 2019–2020.
The study collected data for the blood group and its subtype, which was registered while they were admitted for treatment. Table 2 shows the blood group of patients and their parents. As seen in the table, the proportions of A positive (27%), B positive (27%), and O positive (25%) were almost equally distributed among patients, while the blood groups of AB positive (10%), O negative (4%), A negative (2%), and AB negative (0.2%) were lower compare to former ones. In addition, the subgroups of blood were also reviewed and analyzed, which is depicted in Figure 2. According to the graph, the highest proportion of blood subgroups were recorded for subgroup e+ (95%), subgroup c+ (92%), subgroup C+ (87%), and subgroup E+ (60%). Although, the subgroups of C− (10%), c− (6%), and e− (2%) were in the lower proportions.
| Variables | Patients | Father | Mother | |||
|---|---|---|---|---|---|---|
| Blood groups | n | % | n | % | n | % |
| A− | 7 | 2 | 7 | 2 | 6 | 2 |
| A+ | 110 | 27 | 101 | 27 | 111 | 31 |
| AB− | 1 | 0 | 3 | 1 | 0 | 0 |
| AB+ | 43 | 11 | 35 | 9 | 23 | 6 |
| B− | 16 | 4 | 15 | 4 | 8 | 2 |
| B+ | 111 | 27 | 111 | 30 | 97 | 27 |
| O− | 16 | 4 | 5 | 1 | 7 | 2 |
| O+ | 105 | 26 | 97 | 26 | 105 | 29 |
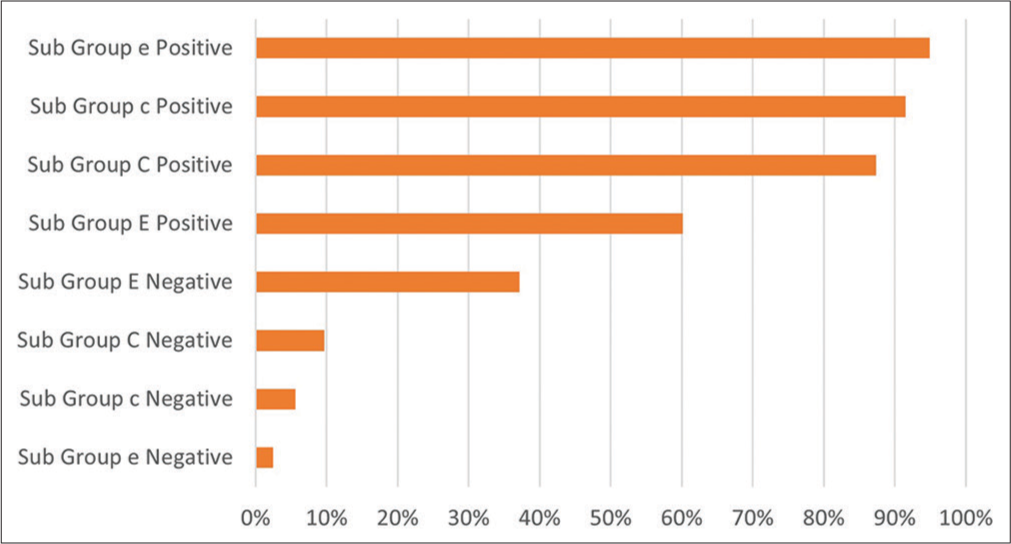
- Proportion of blood subgroups among thalassemia patients in the central blood bank during 2019–2020.
After follow-up with phone calls, a total of 261 (63.35%) responded, and the rest were not connected. Just one patient in the registry has lost his life, while 49 (11.89%) respondents mentioned that they had been witness to the death of other children of their families due to thalassemia. In response to a question about the age of patients when the problem was identified, the family reflected differently based on the gender of the patient. These differences can be seen in Figure 3. However, as a whole, one-third of patients, 76 (29.23%), started to suffer the problem in the 1st months of their life, 135 (51.92%) between 1 and 7 months, and 49 (18.85%) after 7 months of life.
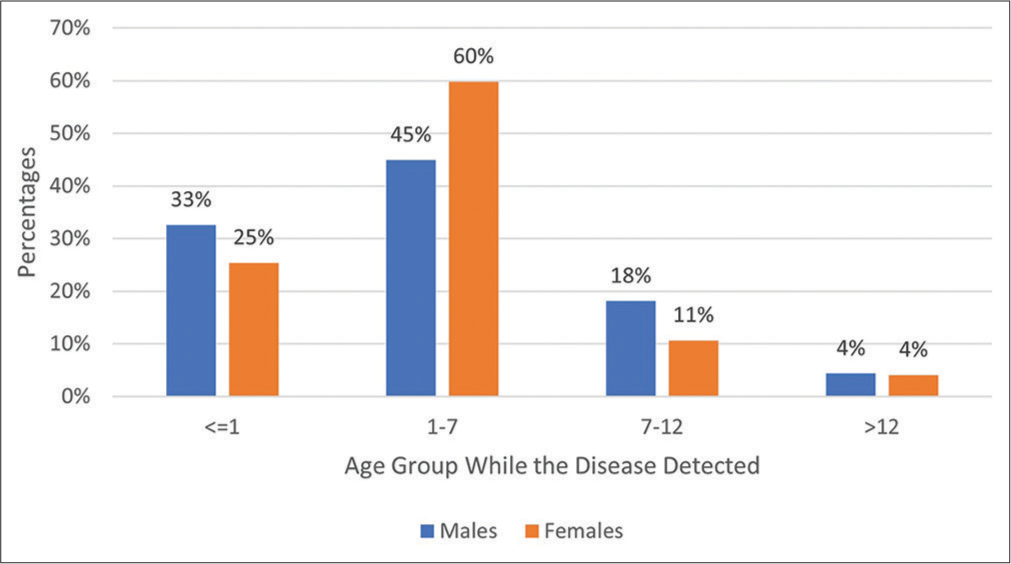
- Age (in months) of diagnosis of thalassemia by differentiation of sex among patients in the central blood bank, 2019–2020.
Of those who responded to phone calls, 18 (6.92%) got operated surgically due to thalassemia, and 181 (69.62%) mentioned that they are using the iron chelating agent (Deferoxamine) for treatment. However, the agent was not available in the center, and they were obliged to purchase it from the private market. In total, 66 (25.88%) replied that they were able to purchase the injections, while others were in low economic status and unable to afford the treatment. Half of the respondents (52%) mentioned that they had approached other centers within and out of the country for treatment of their patients. It should be mentioned that those who were registered in this center were under management for blood transfusion and treatment with chelating agents.
DISCUSSION
The findings of the study showed that the majority of thalassemia cases are major, which are diagnosed and under management, while the prevalence of other cases, including trait or minor and intermedia, which do not require institutional treatment, are not reported. Probably that is due to low seeking care behavior by families who have such genetic disorders. The burden of thalassemia should have been much more than recorded in Afghanistan because with a global frequency of 1.5%. Globally, there is an estimate that more than 300,000 babies are born annually, and 100 million are carriers of thalassemia.[1,5] The study showed slightly more proportions (54.61%) of males with a male-to-female ratio of 1.2:1. This finding is similar to studies carried out in Yemen, Bangladesh, and India.[27-29] The age of patients was clustered around 7 years with no significant difference between boys and girls. This finding is consistent with another study conducted in Yemen[27] and dissimilar to the study conducted in India.[29] Nonetheless, a higher average age of thalassemia patients was reported from UAE, Hong Kong, and Iran.[1-32] More than 90% of patients showed clinical signs and symptoms in the 1st year of their life, which is higher than studies conducted elsewhere and published in the literature.[33,34] This high detection of cases in the 1st year of life is a good sign for taking early measures for management.
We found a high proportion of consanguinity marriage among parents of thalassemia patients, which can be explained by the fact that the disease is transmitted in an autosomal recessive manner and a high consanguinity rate is a distinguishing nature of the problem. In the same way, positive parent consanguinity was found in around two-thirds of cases in Yemen,[27] India,[35] and Iran.[36] The patients who were registered for treatment of blood transfusion and chelation therapy, despite of provision of blood by the center and their relatives, they took deferoxamine from the market. Two-thirds of families of patients registered in the NBB mentioned that they have no economic ability to afford the chelating agents. These findings are similar to other studies.[27,37,38] High level of mortality was reported by families in follow-up by phone calls. Such reports are available in published studies in the literature as well.[39,40] More than 90% of diagnosed patients were thalassemia major, which is consistent with studies in Pakistan.[41]
This finding of this study will open the way for working more and doing further studies regarding genetic blood disorders in Afghanistan. However, our findings were limited to data recorded in registries in the NBB. Despite affecting high morbidity and mortality, the disease could have a considerable level of psychological and economic burden. Hence, the provision of information and acting in this regard is very necessary for the health system. We were bound to registries, so some variables not included in the registry were missed. Another limitation of our study was the difficulty in contacting some patients’ families by phone, probably due to not using or changing the phone and, the unstable situation of the country, and low education and low awareness of the clinical situation of their children. Low awareness, low education, and low capacity of the government to manage the cases are the great reasons for the adverse effects. The establishment of more equipped centers and raising awareness of the disease should be given high priority. As the majority cannot afford to purchase the chelating agent, so the provision of free iron chelating therapy in a sufficient and regular manner for all patients is essential. Donors, influential and rich people, as well as charity organizations should be encouraged to support the thalassemia centers financially. Preventive measures such as health education, enhancing awareness, genetic counseling, and early prenatal diagnosis are measures that can contribute to reducing the incidence of the disease, and through that way it will decrease the social and economic burden on families. Cousin marriage should also be raised as a public health problem and should be discouraged considering its social health and religious reasons.
CONCLUSION
There are many competing health priorities in Afghanistan, being faced with challenges, including the worse status of mother and child health, nutrition, and communicable and non-communicable diseases. Furthermore, very less published information is available about blood genetic disorders, including thalassemia, in Afghanistan, so the findings of this descriptive study are of value for all partners to plan and make interventions for the improvement of the situation. Given the high level of poverty and low socioeconomic status, blood disorders, including thalassemias, are required to be taken seriously.
Acknowledgment
I would like to thank Dr. Nadir Baha the manager of thalassemia section for facilitation the procedure of study in NBB and also Ms Nooria Saeedi research officer in ANPHI for accessing and data entry of the patient records.
Data availability
As the data have been extracted from registry of the NBB available as official documents of this department, if there is a need to have access to these data, it will require some official steps which are required by the government. The corresponding author will facilitate access to the original data if such a request is coming.
Ethical approval
The research/study was approved by the Institutional Review Board at Afghanistan National Public Health Institute, number 12190110, dated 3rd December 2019.
Declaration of patient consent
Patient’s consent was not required as there are no patients in this study.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480-7.
- [CrossRef] [PubMed] [Google Scholar]
- Thalassemia. National Center on birth defects and developmental disabilities. Available from: https://www.cdc.gov/ncbddd/thalassemia/facts.html [Last accessed on 2019 Apr 23]
- [Google Scholar]
- Pathophysiology of thalassemia. Baillieres Clin Haematol. 1998;11:127-46.
- [CrossRef] [PubMed] [Google Scholar]
- Role of deferiprone in chelation therapy for iron overload. Blood. 2003;102:17-24.
- [CrossRef] [PubMed] [Google Scholar]
- Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders. Expert Rev Hematol. 2010;3:103-17.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology In: Galanello R, Eleftheriou A, Traeger-Synodinos J, Petrou M, Angastiniotis M, Galanello R, eds. Prevention of thalassaemias and other haemoglobin disorders. Vol 1. Nicosia, Cyprus: Thalassaemia International Federation Publication; 2005. p. :10-3.
- [Google Scholar]
- The thalassaemia syndrome. (4th). Oxford: Blacwell Scientific Publications; 2001.
- [CrossRef] [Google Scholar]
- Beta-thalassaemia in the immigrant and non-immigrant German populations. Br J Haematol. 1997;97:266-72.
- [CrossRef] [PubMed] [Google Scholar]
- Thalassemia prevalence. Available from: http://www.ironhealthalliance.com/disease-states/thalassemia/epidemiology-and-pathophysiology.jsp [Last accessed on 2024 May 17]
- [Google Scholar]
- Congenital anomalies fact sheet. 2016. Geneva: World Health Organization; Available from: http://www.who.int/mediacentre/factsheets/fs370/en [Last accessed on 2017 Mar 30]
- [Google Scholar]
- Global report on birth defects: The hidden toll of dying and disabled children. 2006. New York: March of Dimes Birth Defects Foundation; Available from: http://www.marchofdimes.org/materials/global-report-on-birth-defects-the-hidden-toll-of-dying-and-disabled-children-full-report.pdf [Last accessed on 2017 Mar 30]
- [Google Scholar]
- Prevalence of heterozygous-thalassemia in the Northern areas of Pakistan. J Pak Med Assoc. 1992;42:32-4.
- [Google Scholar]
- An approach for the prevention of thalassemia in Pakistan. 1998. PhD Thesis. University of London. Available from: http://discovery.ucl.ac.uk/1317916/1/299931.pdf [Last accessed on 2024 May 17]
- [Google Scholar]
- Clinical and hematological picture of multi-transfused thalassemia major patients at a center in Pakistan. J Islam Int Med Col. 2018;13:52-6.
- [Google Scholar]
- Thalassemia in Iran in last twenty years: The carrier rates and the births trend. IJBC. 2013;6:11-8.
- [Google Scholar]
- Frequency of b thalassemia trait and other hemoglobinopathies in Northern and Western India. Indian J Hum Genet. 2010;16:16-25.
- [CrossRef] [PubMed] [Google Scholar]
- Results of programs for antenatal detection of thalassemia in reducing the incidence of the disorder. Blood Rev. 1987;1:169-76.
- [CrossRef] [PubMed] [Google Scholar]
- Burden of thalassemia in India: The road map for control. Pediatr Hematol Oncol J. 2017;2:79-84.
- [CrossRef] [Google Scholar]
- 1,500 children suffering from thalassemia in Nangarhar. Pajjwak News. Health. Available from: https://www.pajhwok.com/en/health [Last accessed on 2024 May 17]
- [Google Scholar]
- β-thalassemia carriers in Afghanistan: A prevalence estimate. Ann Biol Clin. 2013;71:503-4.
- [CrossRef] [PubMed] [Google Scholar]
- Carrier screening for thalassemia and hemoglobinopathies in Canada. Joint SOGCCCMG clinical practice guideline. J Obstet Gynaecol Can. 2008;30:950-9.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and genetic analysis of α-and β-thalassemia and sickle cell anemia in Southwest Iran. J Epidemiol Glob Health. 2018;8:189-95.
- [CrossRef] [PubMed] [Google Scholar]
- What is thalassemia? Available from: http://www.thalassemiaindia.org/what-is-thalassemia.aspx# [Last accessed on 2024 May 17]
- [Google Scholar]
- Thalassemia and other haemoglobinopathies. World Health Organization Resolutions. EB118R1 and WHA5920 2006
- [Google Scholar]
- The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331-6.
- [CrossRef] [PubMed] [Google Scholar]
- Pattern and clinical profile of thalassemia among pediatric patients attending the Yemeni Society Centers for Thalassemia and Genetic Blood Disorders in Yemen. Sci J Al-Azhar Med Faculty Girls. 2017;1:43.
- [CrossRef] [Google Scholar]
- Thalassemias in South Asia: Clinical lessons learnt from Bangladesh. Orphanet J Rare Dis. 2017;12:93.
- [CrossRef] [PubMed] [Google Scholar]
- Chelation status and clinical profile of thalassemic children attending paediatric clinics. Indian J Appl Res. 2016;6:276-8.
- [Google Scholar]
- Prevalence of iron overload complications among patients with bthalassemia major treated at Dubai Thalassemia Centre. Ann Saudi Med. 2013;33:18-21.
- [CrossRef] [PubMed] [Google Scholar]
- Morbidity and mortality patterns of thalassemia major patient in pediatric department of three regional hospital: Retrospective study. Hong Kong Med J. 2002;8:255-60.
- [Google Scholar]
- Metabolic and endocrinologic complications in beta-thalassemia major: A multicenter study in Tehran. BMC Endocr Disord. 2003;3:4.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of consanguinity on screening for thalassemia. N Engl J Med. 2000;347:1200-2.
- [CrossRef] [PubMed] [Google Scholar]
- The clinical approach to thalassemia New York: Grune and Stratton; 1984. p. :151-74.
- [Google Scholar]
- A study on endocrine dysfunction in thalassaemia. J Indian Med Assoc. 2008;106:655-9.
- [Google Scholar]
- Survival analysis and its associated factors of beta thalassemia major in Hamadan Province. Iran J Med Sci. 2015;40:233-9.
- [Google Scholar]
- Study on effectiveness of transfusion program in thalassemia major patients receiving multiple blood transfusions at a transfusion centre in Western India. Asian J Transfus Sci. 2010;4:94-8.
- [CrossRef] [PubMed] [Google Scholar]
- Ethical issues and risk/benefit assessment of iron chelation therapy: advances with deferiprone/deferoxamine combinations and concerns about the safety, efficacy and costs of deferasirox. Hemoglobin. 2008;32:1-15.
- [CrossRef] [PubMed] [Google Scholar]
- Heart failure in beta thalassemia syndromes: A decade of progress. Am J Med. 2005;18:957-67.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiac disease-free survival in patients with thalassemia major treated with subcutaneous deferoxamine. An update of the Toronto cohort. Ann N Y Acad Sci. 1990;612:585-6.
- [CrossRef] [Google Scholar]
- A clinico-epidemiological study of thalassemia cases in India. J Nat Sci Biol Med. 2018;9:236-41.
- [CrossRef] [Google Scholar]




