
Translate this page into:
Exploring the relationship between thalassemia and bone health: A clinicopathological analysis

*Corresponding author: Arijit Das, Department of Medicine, Assam Medical College and Hospital, Dibrugarh, Assam, India. darijit1992@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Das A, Dutta A, Taye P, Sharma A. Exploring the relationship between thalassemia and bone health: A clinicopathological analysis. J Hematol Allied Sci. 2023;3:115-9. doi: 10.25259/JHAS_20_2023
Abstract
Objectives:
To study the bone mineral density in patients with thalassemia. Thalassemia is a genetic disorder characterized by reduced synthesis of the globin chain, leading to decreased hemoglobin levels. Blood transfusion therapy is the primary treatment, but it can cause iron overload and other factors that increase the risk of low bone mineral density. Despite this, there is limited research on thalassemia patients in India, particularly in the North Eastern region, with a focus on bone mineral density.
Material and Methods:
We conducted a hospital-based case-control study in the Department of Medicine at Assam Medical College and Hospital, Dibrugarh, from June 1, 2020, to May 31, 2021. We have collected data from 51 cases and 51 healthy controls and analyzed it (SPSS for Windows, version 21.0 Chicago, SPSS Inc.) and Microsoft Excel 2010. Bone mineral density was measured by Dual-Energy X-ray Absorptiometry (DEXA) (Lunar Prodigy Advance DEXA (GE Healthcare, Madison, WI, USA).
Results:
Our study also showed that thalassemia patients had a 3.775 times higher risk of low bone mineral density at the lumbar vertebra and a 4.0421 times higher risk at the femur neck than the normal healthy population. The mean BMD at the lumbar spine and femur neck were lower for cases than controls, with the difference at the femoral neck being statistically significant.
Conclusion:
Our study highlights that more than half of thalassemia patients have low bone mineral density, with the femoral neck and lumbar neck being more affected than normal age and sex-matched controls. These findings underscore the importance of monitoring bone health in thalassemia patients and implementing appropriate interventions to prevent or manage low bone mineral density.
Keywords
Thalassemia
Bone mineral density
Osteoporosis
Osteopenia

INTRODUCTION
Thalassemia is a group of recessively inherited disorders of reduced hemoglobin (Hb) synthesis due to β-globin chain mutation. The thalassemia is broadly divided into α-thalassemia, β-thalassemia, or thalassemic variants (structurally abnormal Hb is associated with coinherited thalassemia phenotypes, e.g., hemoglobin E [HbE]).[1]
The homozygous state of the disease results in severe anemia, ultimately requiring more blood transfusion. The current management of thalassemia major, which includes regular transfusion therapy and concurrent iron chelation therapy, has significantly improved the patient’s life expectancy. Unfortunately, this increased longevity has been at the cost of increased risk of complications. Thalassemia is associated with multiple complications due to iron overload involving various organs such as the pituitary, thyroid, pancreas, gonads, parathyroid, and bone.[2]
One of the most common endocrine complications is low bone mass. It has emerged as a major challenge affecting both children and adolescents with transfusion-dependent thalassemia despite adequate blood transfusion and iron therapy. Various factors lead to low bone mass, which includes iron deposition, expansion of the medulla, hormone deficiency, rapid bone turnover, calcium-phosphorus imbalance, and hypoxia.[3] Low bone mass leads to bone deformity, pain, marrow expansion, osteopenia, osteoporosis, and fractures.[4-7] It has also been observed that low bone mass is highly prevalent among children who are transfusion-dependent. However only few studies have shown that nontransfusion-dependent thalassemia patients are also likely to develop low bone mass later in life.
The prevalence of low bone density in thalassemia has been well documented. Still, studies examining the changes in bone mineral density (BMD) in patients with thalassemia are very limited in number in India, mostly in the North East region. Hence, this study was conducted to have a better understanding of the prevalence of low bone mass among adolescents with thalassemia and related various clinical factors. The findings of this study will broaden the understanding of this disorder, which will lead to early diagnosis and effective management.
MATERIAL AND METHODS
This hospital-based case–control study aimed to describe the clinicopathological phenotype of patients with thalassemia and assess BMD using bone densitometry. The study was conducted in the Department of Medicine, Assam Medical College and Hospital, Dibrugarh, for one year from June 1, 2020, to May 31, 2021, after obtaining ethical clearance from the Institutional Ethics Committee (Human) of Assam Medical College and Hospital, Dibrugarh, and informed consent from the study participants.
Patients diagnosed with thalassemia aged over 12 years were selected after screening from those attending the outpatient department of Medicine or the Hematology clinic and those admitted as inpatients at the Department of Medicine, Assam Medical College, Dibrugarh. A total of 51 cases were included in the study, and gender-, age-, and ethnicity-matched healthy control subjects were randomly chosen among the workers, staff, and attendants of the patient.
Anthropometry was performed, including height and weight measurements. BMD was measured by dual-energy X-ray absorptiometry (DEXA) using Lunar Prodigy Advance DEXA (GE Healthcare, Madison, WI, USA) at the lumbar spine (LS) (L1–L4) and femoral neck (FN). The results obtained for each subject were recorded and expressed as BMD of the LS and FN as an absolute value (gram/cm2). Osteopenia was defined as a T score between −1 and −2.5, while osteoporosis was defined as a T score below −2.5. In this study, osteoporosis was considered as a Z score ≤ −2.5 standard deviation (S.D) and osteopenia as a Z score between −1 and −2.5 S.D.
Osteopenia is defined as a T score between −1 and −2.5, while osteoporosis is defined as a T score below −2.5.120. The World Health Organization’s definition of osteoporosis applies only to adult women and not to men or children. The main reason for this is that the relationship between bone mass and fracture risk is not as clear in men as it is in postmenopausal women, and it is not established at all in children.
Z scores are preferred in females before menopause and in males <50 years of age.[8] As a result, we expressed BMD values as Z score; a Z score represents the number of standard deviations above or below the age- and sex-matched mean reference value. Few studies have categorized osteopenia and osteoporosis based on the Z Score.[9-11] Thus, in our study, we consider osteoporosis as a Z score ≤ −2.5 S.D and osteopenia as a Z score between −1 and −2.5 S.D.
The statistical analysis of data was performed using the computer program Statistical Package for the Social Sciences (SPSS for Windows, version 21.0 Chicago, SPSS Inc.) and Microsoft Excel 2010. Results on continuous measurements presented as mean ± standard deviation were compared using an independent t-test, and discrete data were expressed as numbers (%) and analyzed using the Chi-square test. The statistical significance was fixed at a 5% level (P < 0.05).
RESULTS
In our study, a total of 51 cases and 51 controls, both age- and sex-matched, were selected. Out of 51 cases, 26 were male (50.98%) and 25 were female (49.02%). The male-to-female ratio was 1.04:1.
The mean age of cases in our study was 20.14 ± 9.98 years. The majority, 36 (70.59%), belong to the age group of 13–20 years. Majority of thalassemia patients included in the study, 25 (49.02%) were diagnosed at <3 Years of age, with a mean age at diagnosis being 3.62 + 1.12 years.
In the present study, the majority of the cases, 20 (39.22%), belonged to the Tea Tribe community, followed by the Assamese population 16 (31.37%), and the remaining around 30% of the cases belonged to the community such as Bengali, Bihari, Bodo, and Nepali.
Based on the high-performance liquid chromatography report, out of 51 cases included in our study, 27 (52.94%) cases had HbE-β-thalassemia, 22 (43.13%) cases had β-thalassemia major, and 2 (3.93%) patients had β-thalassemia intermedia [Figure 1].
In our study, on examination of 51 cases, it was found that generalized weakness and easy fatigability were the most common symptoms in almost all thalassemia patients, followed by dyspnea on exertion. A maximum number of patients developed icterus which was seen in 24 (88.89%) cases of HbE-β-thalassemia and 11 (81.25%) cases of β-thalassemia major. Pallor was invariably present. Splenomegaly was present in 19 (85.18%) cases of HbE-β-thalassemia major and 11 (68.75%) of β-thalassemia major.
Out of 36 cases (70.59%) who were receiving chelation therapy, only 10 (19.61%) were on regular chelation therapy, and 26 (50.98%) were on irregular chelation therapy.
On the measurement of BMD at LS using DEXA scan, out of 51, 37 patients were found to have low BMD; that is, 17 (33.33%) patients had osteopenia, while 20 (39.22%) patients had osteoporosis. Whereas, in comparison to controls, 13 (25.49%) had osteopenia and 8 (15.69%) had osteoporosis [Figure 1]. Again, it was found that the risk of having low BMD at LS is 3.775 times more (odds ratio [OR] - 3.775, P = 0.001) common in thalassemia patients when compared to a normal healthy population [Figures 2 and 3].

- Type of thalassemia based on high-performance liquid chromatography report.
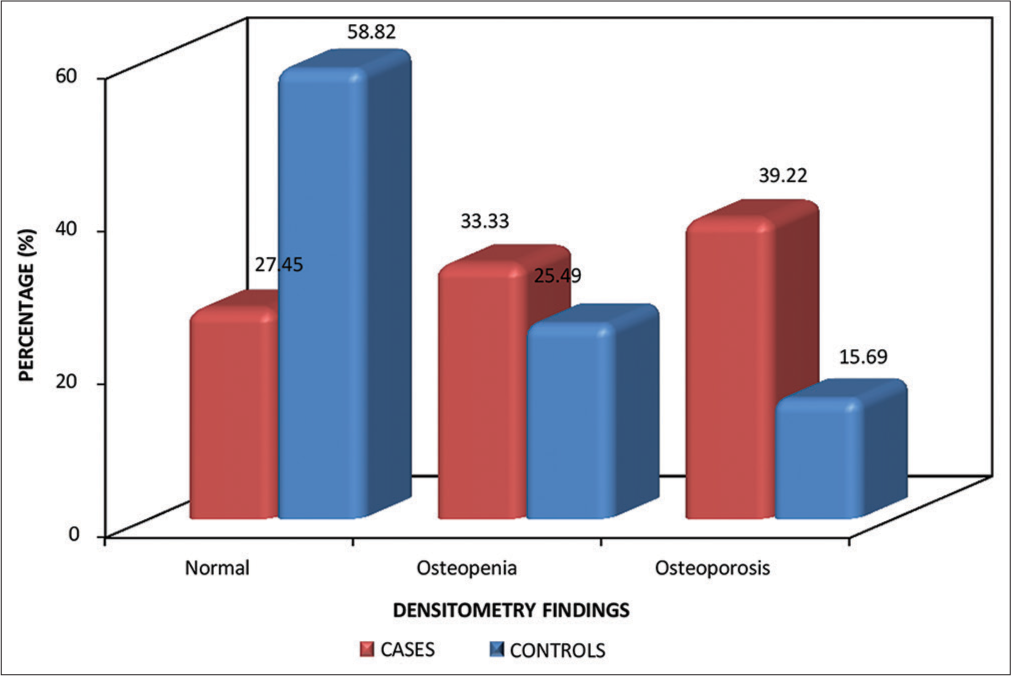
- Bone densitometry findings based on z score at lumbar spine.

- Association between low bone mineral density at lumbar spine with thalassemia.
Similarly, on measurement of BMD at the femur neck using a DEXA scan, out of 51, a total of 32 patients had low BMD; that is, 22 (43.14%) patients had osteopenia, while 10 (19.61%) patients had osteoporosis. Whereas in comparison to controls, 12 (23.53%) had osteopenia and 3 (5.88%) had osteoporosis. In our study, the risk of having low BMD at the femur neck in a thalassemia patient is 4.0421 times more (OR – 4.0421, P = 0.001) when compared to the normal healthy population.
In our study, the mean BMD at LS in cases was 0.852 ± 0.129 g/cm2, whereas in the control group, the mean BMD at LS was 0.879 ± 0.286 g/cm2. Although the BMD at the LS in cases was less than that of BMD in controls as compared to the cases, it was not statistically significant. The mean BMD at the FN in cases was 0.887 ± 0.185 g/cm2, whereas, in controls, the mean BMD at the FN was 0.943 ± 0.157 g/cm2; however, this difference was statistically significant (P = 0.048).
DISCUSSION
This is the first study done in the Northeastern part of India focused on studying the clinical profile of the patient with thalassemia with a special preference for BMD. The results show a high prevalence of low BMD in adolescents with thalassemia in comparison to normal healthy controls.
In our study, most of the patients presented had generalized weakness and fatigue followed by dyspnea on exertion. Other signs and symptoms observed are pallor, icterus, splenomegaly, and hepatomegaly. Pallor and icterus were seen in almost all the cases. Similarly, in the studies conducted by Deb et al. and Saikia Pathak et al., almost all the cases presented with generalized weakness and fatigue.[12,13]
Splenomegaly was present in 85.18% of cases of HbE-β-thalassemia, 68.75% of β-thalassemia major, and none in thalassemia intermedia. Moreover, a mild-to-moderate degree of hepatomegaly is present in 88.89% of cases of HbE-β-thalassemia, 81.25% of β-thalassemia major, and none in thalassemia intermedia. These findings were greater in percentage as compared to the study of Deb et al., which can be due to chronic malaria as most of the patients hail from malaria-endemic areas.[12]
Clinical manifestation can be due to inadequate blood transfusion and irregular chelation therapy. Again, even frequent transfusion can lead to an iron overload state in various organs, leading to hypogonadism, diabetes mellitus, hypothyroidism, hypoparathyroidism, and other endocrine problems, cardiomyopathy, hepatic fibrosis, and cirrhosis, which can define the symptomatology.
The majority of the patients, 36 (70.59%), who were on blood transfusion belong to the younger age group of 13–20. Again, 45.1% of cases received 10 transfusions/year, 45.1% received 10–20 transfusions/year, and 9.8% received more than 20 transfusions/year. In a similar study carried out by Singhal et al., out of 63 thalassemia patients, 2 (3.17%) received <10 transfusions/year, 25 (39.68%) received 10–20 transfusions/year, and 24 (38.09%) received more than 20 transfusions/year.[14] In our study, the mean Hb among cases was 7.74 ± 2.16 g/dL which indicates that majority of the patient in our study were under transfused.
The majority of the patients with thalassemia have limited access to regular and safe blood transfusions. A lack of unpaid voluntary blood donors, a lack of thalassemia awareness, a lack of national blood policies, and fragmented blood services are the various factors that contribute to a significant gap between timely supply and demand for safe blood.
In the present study, out of 36 cases (70.59%) who were receiving chelation therapy, only 10 (19.61%) were on regular chelation therapy, and 26 (50.98%) were on irregular chelation therapy. There is a lack of awareness as well as financial constraints that lead to non-adherence to the chelation regime. Our study findings are in accordance with those of Kataki, Bharati, and Singhal et al.[14,15]
Patients with thalassemia major have a better chance of survival due to regular blood transfusion regimens and early iron chelating therapy. Despite being well-transfused and receiving iron chelation, severe osteoporosis, increased spine trabeculation, and osteopenia with cortical thinning remain serious complications.
Our findings in a group of adolescents with thalassemia show low BMD is a common feature. On the measurement of BMD at LS using a DEXA scan, out of 51, 37 patients were found to have low BMD. That is, 17 (33.33%) patients had osteopenia, while 20 (39.22%) patients had osteoporosis. Whereas in comparison to controls, 13 (25.49%) had osteopenia, and 8 (15.69%) had osteoporosis.
These findings are in concordance with Mahmoodi Nesheli et al., who observed the BMD findings in LS in cases: Normal (17.1%), osteoporosis (48.6%), and osteopenia (34.3%).[16]Another study done by Singh et al. showed similar results at LS: 20% had normal BMD, 37.5% had osteopenia, and 42.5% had osteoporosis.[9] Another study conducted by Shamshirsaz et al. observed similar findings where the prevalence of osteoporosis and osteopenia in the LS was 50.7% and 39.4%, respectively.[10]
At the femur neck, it is seen that 19 (37.25%) cases had normal BMD, whereas in the control group, 36 (70.59%) had normal BMD. Out of 51 cases, 22 (43.14%) had osteopenia; on the other hand, 12 (23.53%) subjects in the control group had osteopenia. Among the cases, 10 (19.61%) had osteoporosis, while, in the control group, 3 (5.88%) had osteoporosis. Between cases and controls, there is a statistically significant difference in BMD status (P < 0.001). Other studies done by Mahmoodi Nesheli et al., Singh et al., and Shamshirsaz et al. have reported similar findings.[9,10,16]
In our study, out of 51 cases, 72.54% (37 out of 51) showed bone involvement at LS in comparison to 41.17% (21 out of 51) in controls. The odds Ratio calculated is 3.775, which validates the risk of having bone involvement at LS in thalassemia patients is 3.775 times more (OR – 3.775, P = 0.001) when compared to a normal healthy population. Similarly, when BMD was calculated at the femur neck and compared to controls, the risk of having bone involvement at the femur neck in thalassemia patients was found to be 4.0421 times more when compared to a normal healthy population.
Hence, from our study, it has become evident that thalassemia patients are more prone to develop osteopenia and osteoporosis as compared to normal populations, which is in concordance with the studies discussed above.
We have seen that despite regular blood transfusions and chelation therapy, patients develop low BMD. This raises the question of multiple risk factors that play a role in BMD, which is different from the normal population. Various other factors, such as abnormal GH-IGF-1 axis, hypogonadism, and vitamin D deficiency, also contribute to low BMD. However, we could not carry out tests such as phosphorus, parathyroid hormone, vitamin D3, and calcium levels due to funding limitations, which would have provided further insights into the topic.
In summary, our study reported the prevalence of low bone mass measured at LS and FN among more than half of the adolescents with thalassemia. We also found an inverse relationship between BMD and pretransfusion Hb levels. Physicians should be alert while treating patients with thalassemia, as effective management and regular follow-up can improve long-term bone health.
It is true that the sample size of this study was relatively small, and further studies with larger sample sizes and longer durations are needed to understand better the factors responsible for BMD changes in thalassemia patients. In addition, it is important to note that DEXA is a two-dimensional measurement that is correlated to body size and bone size, which can lead to deviations in growing populations. Therefore, future studies should consider using three-dimensional imaging techniques to obtain more accurate measurements of BMD in thalassemia patients. Despite these limitations, this study provides valuable insights into the clinical profile of thalassemia patients in the Northeastern part of India. It highlights the need for regular monitoring and management of bone health in these patients.
CONCLUSION
This study provides valuable insights into the clinical profile of thalassemia patients in the North Eastern part of India. This study found that over half of the thalassemia patients had low BMD, with osteoporosis and osteopenia being the most common forms. The FN and LS were more affected in thalassemia patients compared to age- and sex-matched controls. These findings highlight the need for regular monitoring and management of bone health in thalassemia patients to prevent the development of serious complications.
Ethical approval
The research/study is approved by the Institutional Ethics Committee at Assam Medical College and Hospital, number AMC/EC/PG-8842 dated 3rd November 2021.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript, and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Pathophysiology of thalassaemia. Baillieres Clin Haematol. 1998;11:127-46.
- [CrossRef] [PubMed] [Google Scholar]
- Multicentre study on prevalence of endocrine complications in thalassaemia major. Italian Working Group on Endocrine Complications in Non-endocrine Diseases. Clin Endocrinol (Oxf). 1995;42:581-6.
- [CrossRef] [PubMed] [Google Scholar]
- Bone mineral density in thalassemia major patients from antalya, Turkey. Int J Endocrinol. 2012;2012:573298.
- [CrossRef] [PubMed] [Google Scholar]
- Oxidative stress and iron homeostasis: Mechanistic and health aspects. Crit Rev Clin Lab Sci. 2008;45:1-23.
- [CrossRef] [PubMed] [Google Scholar]
- Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202:199-211.
- [CrossRef] [PubMed] [Google Scholar]
- Survival and causes of death in thalassaemia major. Lancet. 1989;2:27-30.
- [CrossRef] [PubMed] [Google Scholar]
- International society for clinical densitometry 2007 adult and pediatric official positions. Bone. 2008;43:1115-21.
- [CrossRef] [PubMed] [Google Scholar]
- Status of 25-hydroxyvitamin D deficiency and effect of vitamin D receptor gene polymorphisms on bone mineral density in thalassemia patients of North India. Hematology. 2012;17:291-6.
- [CrossRef] [PubMed] [Google Scholar]
- Metabolic and endocrinologic complications in beta-thalassemia major: A multicenter study in Tehran. BMC Endocr Disord. 2003;3:4.
- [CrossRef] [PubMed] [Google Scholar]
- Study on effectiveness of transfusion program in thalassemia major patients receiving multiple blood transfusions at a transfusion centre in Western India. Asian J Transfus Sci. 2010;4:94-8.
- [CrossRef] [PubMed] [Google Scholar]
- Clinicohaematological spectrum of haemoglobinopathies a hospital based study. J Evol Med Dent Sci 2015:414934-42.
- [CrossRef] [Google Scholar]
- Disorders of haemoglobin variants in paediatric patients attending in a tertiary care hospital of North East India. Int J Biol Med Res. 2014;5:3841-6.
- [Google Scholar]
- Iron overload and growth of thalassemic patients in Marwar region. Int J Pharm Sci Res. 2012;3:2043-9.
- [Google Scholar]
- Relationship between serum ferritin and endocrinopathies in thalassemic children: A hospital-based study. Indian J Child Health. 2019;6:201-4.
- [CrossRef] [Google Scholar]
- Relation between bone mineral density and serum ferritin levels in patients with thalassemia major. Casp J Pediatr. 2016;2:158-63.
- [Google Scholar]




