
Translate this page into:
Inherited platelet function disorders in children – A diagnostic conundrum solved by multimodality testing

*Corresponding author: Prerna Arora, Department of Pathology (Hematology), Maulana Azad Medical College, New Delhi, Delhi, India. drprernaarora27@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Arora P, Munjal P, Parikh H, Viswanathan GK. Inherited platelet function disorders in children – A diagnostic conundrum solved by multimodality testing. J Hematol Allied Sci. 2025;5:72-6. doi: 10.25259/JHAS_6_2024
Abstract
Inherited platelet function disorders (IPFDs) are an extremely rare cause of bleeding in hematological practice. These disorders have varied clinical presentations and heterogeneous underlying pathologies. The IPFDs remain largely undiagnosed or misdiagnosed due to lack of clinical suspicion, masquerading as other acquired causes of bleeding and unavailability of specialized tests in resource constraint settings. Glanzmann thrombasthenia (GT) and Bernard–Soulier syndrome (BSS) are rare autosomal recessive platelet surface receptor disorders of glycoprotein (GP)IIb/IIIa and GPIb/IX/V, respectively, with an estimated prevalence of 1/1,000,000 individuals. Six children presenting with profuse bleeding were evaluated. Complete clinical details of bleeding history along with family history and history were taken. A complete hemogram and peripheral smear examination were done. Coagulation studies, light transmission aggregometry coupled with flow cytometry (FCM) for platelet GP expression, were done to determine the cause of bleeding. In the present series, five young children were diagnosed as GT from two different families, and one case was diagnosed as BSS in a young female child, which was misdiagnosed as immune thrombocytopenia at presentation. Careful re-evaluation coupled with clinical history and FCM analysis leads to a confirmed diagnosis. The complex and heterogeneous pathogenesis of rare IPFDs continues to challenge clinicians and the diagnostic laboratories that assess patients for potential bleeding disorders. A high index of suspicion coupled with utilizing multi diagnostic modalities in these rare disorders can clinch the correct diagnosis and help in timely management.
Keywords
Bernard–Soulier syndrome
Giant platelet
Glanzmann thrombasthenia
Platelet function disorder
Thrombocytopenia

INTRODUCTION
Inherited platelet function disorders (IPFDs) comprise rare conditions and remain underdiagnosed despite clinically significant bleeding diathesis. These disorders are a heterogeneous group of mucocutaneous bleeding disorders of variable severity, particularly in situations of high hemostatic compromise.[1-3] Glanzmann thrombasthenia (GT) and Bernard–Soulier syndrome (BSS) are rare autosomal recessive platelet surface receptor disorders of glycoprotein (GP)IIb/IIIa and GPIb/IX/V, respectively, with an estimated prevalence of 1/1,000,000 individuals.[4] A higher prevalence has been observed in populations with consanguineous marriages and may be higher than estimated as often these patients go unnoticed for many years and even their entire lives.[5] A high index of clinical suspicion should be kept for these disorders as timely and accurate diagnosis is of great importance not only for preventive care but also helps in appropriate, timely management and avoiding unnecessary treatment with steroids or even invasive procedures like splenectomy. We, hereby, present six young children who remained undiagnosed previously for a long time and were confirmed with a diagnosis of IPFDs at our institute.
Six children from three families presenting with a clinical history of bleeding were evaluated for a complete workup. Informed consent and assent were taken from all the patients. Complete clinical details of bleeding history along with family history and history of bleeding were taken. The bleeding symptoms were scored as per the International Society of Thrombosis and Hemostasis–bleeding assessment tool into significant and not significant by a total of all the individual scores drawn for the final bleeding score (BS). The BS of ≥3 was considered as abnormal. A complete hemogram and peripheral smear examination were done. The bleeding time and clot retraction tests were not done on any patient. Prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen level, thrombin time (TT), and urea clot solubility test (CST) were done in all cases. The other tests done to supplement the diagnosis were von Willebrand factor (vWF) antigen level by immunoturbidimetry (STAGO – STA compact) and ristocetin cofactor activity. Light transmission aggregometry (LTA) to assess platelet function was performed on a platelet aggregation analyzer, AggRAM, by Helena laboratories. Agonists used were adenosine diphosphate (ADP) (20 μM/L), epinephrine (10μM/L), collagen (10 μg/mL), and ristocetin (1.5 mg/mL). Platelet flow cytometry (FCM) was performed on 5 μL citrated anti-coagulated peripheral blood within two h of sample collection on 3-laser, 8-color BD FACS Canto II flow cytometer (BD Biosciences, USA) using pretitrated amounts of CD42a, CD42b, CD61, and CD41.
CASE SERIES
Case 1 and Case 2
Two siblings of 10 years, male and five years female, reported with the chief complaint of bleeding gums. The elder sibling had a history of spontaneous bleeding for the past ten days, while the younger sibling had bleeding for five days. The BS was 9 and 7, respectively. The elder sibling was second in order of three children born of third-degree consanguineous marriage. The eldest child of the family was 13 years old with no significant bleeding history. They gave no family history of any bleeding disorder. The birth history was uneventful. On examination, both siblings had purpuric patches on their arms and legs in various stages of healing. Past hematological evaluation of both siblings revealed recurrent episodes of epistaxis, bleeding gums, and easy bruisability with no evidence of trauma. The symptoms started at the age of 2 years.
In addition, there was a history of multiple packed red cell and platelet transfusions in the elder sibling, while the younger sibling at the time of presentation required one unit of PRBC transfusion. Hematological investigations in siblings revealed severe microcytic hypochromic anemia with normal platelet count and morphology. Coagulation workup revealed normal PT, APTT, TT, and fibrinogen and urea CST. vWF antigen and ristocetin cofactor activity were within normal limits. Platelet function test by LTA revealed the absence of platelet response with ADP, epinephrine, and collagen, while the response with ristocetin (1.5 mg/mL) was present with characteristic aggregation and disaggregation pattern suggestive of GT [Figure 1a]. Further, confirmation of the diagnosis was done by FCM, which revealed the complete absence of platelet GP expression by GPIIb (CD41) and GPIIIa (CD61). A final diagnosis of Type I GT (<5% expression) was, thus, made in both siblings [Figure 1b].

- (a) Light transmission aggregometry chromatograph on platelet rich plasma (PRP) standardized with platelet poor plasma (PPP) with concentration as mentioned (Conc) showing absent platelet aggregation to reagents (Reag) adenosine diphosphate channel 1, collagen channel 2 and epinephrine channel 3, and response to ristocetin channel 4 (1.5 mg/mL) is present with characteristic aggregation and disaggregation pattern (brown colored) suggestive of Glanzmann thrombasthenia (GT) and (b) flow cytometry gating platelets on SSC (side scatter) and FSC (forward scatter)– absent expression of platelet glycoproteins (GPs) CD41 fluorescein isothiocyanate (FITC) (GpIIb) and CD61 phycoerythrin (PE) (GpIIIa) suggestive of Type I GT (<5% expression), normal CD42a PerCP-Cy 5.5 (GpIb-IX-V), and CD42b allophycocyanin (APC- A) (GpIb-IX-V) expression. Chnl: Channel, Reag: Reagent, Conc: Concentrationion, SSC: Side scatter area, FITC: Flourocein isothiocynate, FSC: Forward scatter area, APC A: allophycocyanin
Case 3, 4 and 5
Three siblings born out of a consanguineous marriage, aged ten years male, six years female, and 3.5 years male, presented with recurrent epistaxis and gum bleeding since childhood. The median age at first symptoms of bleeding was two years in all the siblings. There was no history of trauma with an uneventful birth history. Both the parents did not have any bleeding manifestations. A history of PRBC transfusions was found in both the younger siblings; however, the eldest sibling did not require any transfusion and was given oral antifibrinolytics previously. Blood investigation revealed hemoglobin (Hb) of 8 g/dL in the eldest sibling, while the younger siblings had severe microcytic hypochromic anemia with Hb of 4 g/dL. A smear examination revealed normal platelet count and morphology in all the cases. Screening coagulogram, urea CST, vWF antigen, and Ristocetin cofactor activity were within normal limits. Platelet function test by LTA in all three siblings revealed the absence of platelet response with ADP, epinephrine, and collagen, while the response with ristocetin (1.5 mg/mL) was present and, hence, was diagnosed to have GT [Figure 1a]. FCM in all three cases was done as the eldest sibling had a varied presentation and minor bleeding as compared to the younger siblings. Complete absence of platelet GP CD41 and CD 61 was found in the younger siblings, and they were considered to have Type I GT (<5% expression), while the eldest sibling had a Type III GT variant pattern with normal expression of platelet GP expression but characteristic pattern on platelet LTA. These findings highlight the heterogeneity of this rare disorder with respect to the nature and severity of bleeding. Due to further limitations and a lack of infrastructure at our center, the genetic landscape, especially of Type III GT, could not be undertaken.
Case 6
An 8-year-old female presented to the emergency department complaining of five episodes of spontaneous profuse bilateral epistaxis over the past ten days. There was a history of 2 units of random donor platelet transfusion from outside before she presented to our center. She gave no history of trauma or bleeding from any other site. On examination, no petechiae, bruises, rashes, or organomegaly were found. Her hemogram revealed Hb of 6 g/dL with a platelet count of 4000/uL. A peripheral smear revealed microcytic hypochromic anemia. Viral serology and autoimmune workup were negative. Clinical suspicion of immune thrombocytopenia (ITP) was considered, and bone marrow was performed, which revealed megakaryocytic preponderance with focal clustering. Combined with clinical history and bone marrow findings, the diagnosis of ITP was considered. The patient was given supportive treatment and intravenous immunoglobulin IVIG therapy; however, her bleeding persisted despite treatment. On eliciting a detailed history, there was a prior history of emergency hospital admission at five years of age with a similar episode of profuse bleeding, which required nasal packing and administration of oral tranexamic acid. Profuse epistaxis also leads to hypovolemic shock, requiring multiple platelet and PRBC support along with cryoprecipitate. On questioning her growth and developmental parameters, the parents stated that she achieved developmental milestones as expected and performed well in school. Antenatal and birth history were uneventful, with no abnormal bleeding. Her parent’s marriage was consanguineous, with both parents having no history of bleeding.
On re-review of the peripheral smears, giant forms of platelets were observed along with a high mean platelet volume of 16 fl [Figure 2]. There was no platelet granular abnormality or any inclusions in leukocytes. Her coagulogram revealed normal PT, APTT, fibrinogen, urea CST, and a vWF study. Based on the clinical history, unresponsiveness to ITP treatment, and coagulogram findings, the possibility of BSS was suspected. Platelet aggregometry by LTA could not be performed in view of her low platelet counts. FCM analysis revealed a complete absence of platelet GP expression with CD42a and CD42b [Figure 2]. The patient was given a final diagnosis of BSS, and family studies were advised.
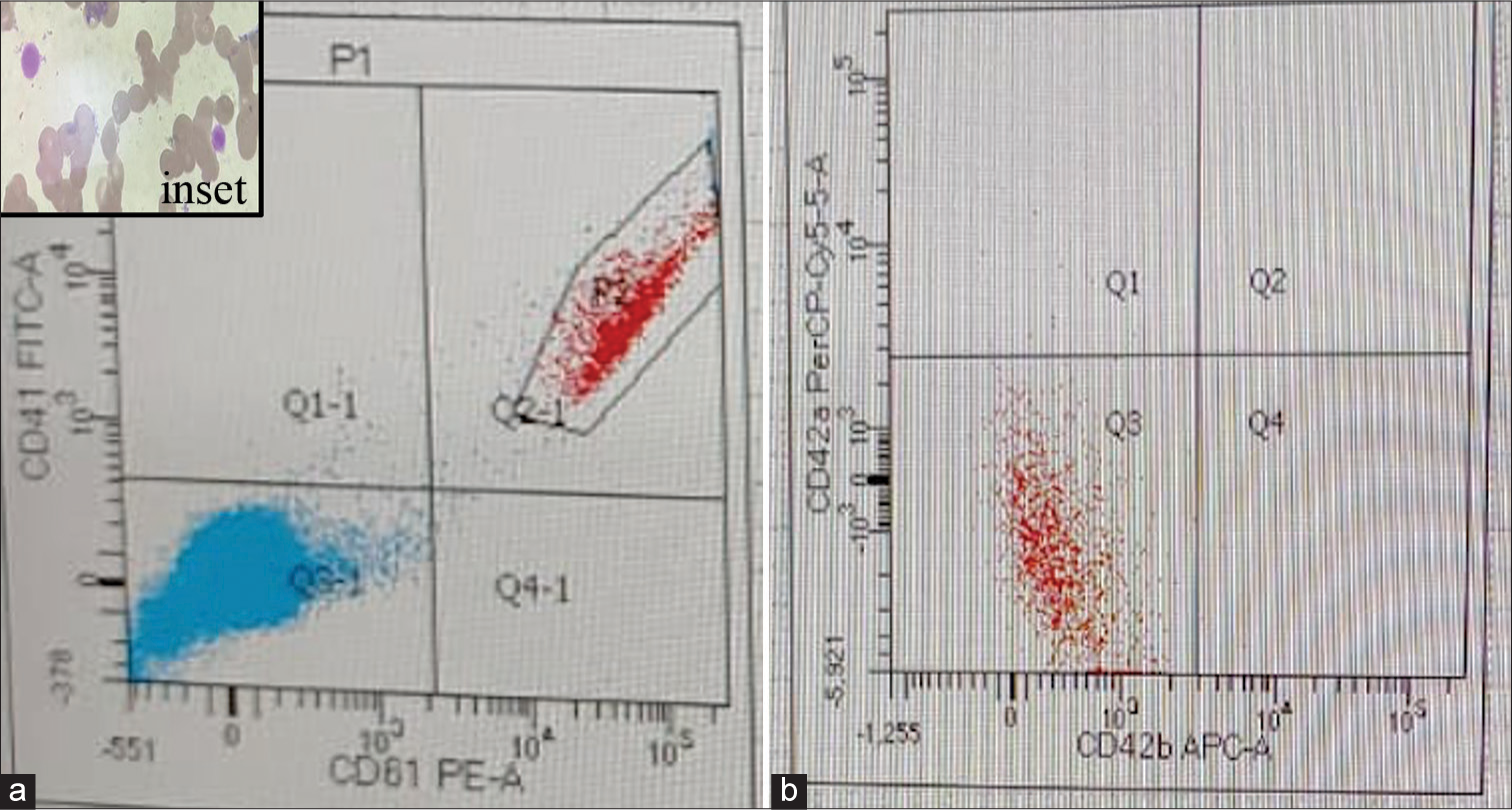
- (a) Flow cytometry expression on gated paltelets (P2, Quadrant Q2-1) revealing normal expression by CD 41 (Gp IIb) FITC and CD61 (Gp III a) PE and (b) revealing absent expression by CD42a (Gp Ib -IX -V) and CD42b (Gp Ib -IX -V)(Q3 quadrant). Inset: Geimsa stained peripheral smear revealing giant platelets.
DISCUSSION
In the present study, patients with inherited PFD went underdiagnosed for a long time despite significant childhood bleeding diathesis. Detailed clinical history along with BS and utilizing multidiagnostic modality leads to the confirmed diagnosis in our patients. GT and BSS are very rare disorders, but they are particularly important for their severity and their biological characteristics.[6] Both disorders show a higher prevalence in populations with consanguineous marriages.[7] All patients in the present series were born to consanguineous parents. GT shows normal platelet count and morphology but abnormal platelet functions due to a defective GP IIb-IIIa complex. Purpura, epistaxis, gingival hemorrhage, and menorrhagia are the recurring features seen in GT.[8] All patients in our series had epistaxis, gum bleeds, and purpuric manifestations, with four out of five patients requiring transfusion support. There are three groups of GT. Patients with Type I have <5% of GPIIb/IIIa; Type II have 5–20% of the GPIIb/IIIa; and Type III (variant type) shows a normal expression of a dysfunctional GpIIb/IIIa. Type I GT is the most prevalent and severe.[9] In our case series, four children were diagnosed as Type 1 GT, while one sibling of the same family was Type III GT (variant) with minor bleeding manifestations. The potential differential diagnosis of variant GT includes autosomal dominant GT-like syndromes and recently described leukocyte adhesion deficiency Type III defects attributed to pathogenic mutations in the FERMT3 gene and such cases present with GT-like bleeding pattern with normal expression of integrins on FCM. Autosomal dominant GT-like syndromes present with platelet anisocytosis and macrothrombocytopenia. However, in the present case of variant GT, macrothrombocytopenia was not found, and diagnosis in other siblings of the same family revealed Type I GT; hence, the possibility of variant GT was considered with minor bleeding presentation. The literature database has found more than 236 mutations in ITGA2B and 140 variants in ITGB3 leading to truncating, missense, single nucleotide variants, splicing defects, deletions, insertions, and inversions in patients of GT, which might explain phenotypic diversity in members of the same family. Although a review of the literature mentions little connection between the seriousness of the disease and the subtypes of GT, we found the milder presentation of Type III GT requiring no transfusions till the time of presentation at ten years of age. Sequence analysis might explain the genotype-phenotype spectrum in such cases. Genotyping for mutations in ITGA2B, ITGB3, and other mutations outside of those causing GT would be beneficial to differentiate from GT, like bleeding disorders, as well as for precise genetic diagnosis and in determining the phenotype variability and heterozygous carriers in families. BSS is a quantitative or qualitative platelet defect due to a lack of GpIb-IX-V complex, resulting in a defective adhesion of the platelet to the subendothelium. The platelet counts in BSS can vary from extremely low (<30×109/L) to near normal (100–200×109/L) and can fluctuate. BSS is extremely rare, and the actual rate may be higher due to the misdiagnosis or underreporting of this condition.[10] BSS can be misdiagnosed as ITP based on clinical manifestations coupled with thrombocytopenia on hemogram. The case of BSS in the present study was also considered as ITP initially, but due to the lack of response to ITP treatment, disproportionate degree of bleed, clinical suspicion, and review of past bleeding history along with giant platelets on smear evaluation coupled with FCM clinched the correct diagnosis. It is thus important to differentiate BSS from other inherited bleeding disorders and ITP so as to prevent any unnecessary treatment with steroids and invasive procedures like splenectomy in unresponsive cases.[11] Mucocutaneous bleeding is a common presenting symptom in these patients; however, the patient in the present case had a prior presentation of hypovolemic shock due to profuse bleeding, signifying the importance of diagnosing this rare entity timely. Due to the limitation of thrombocytopenia in our case, platelet aggregation by LTA could not be performed, and FCM confirmed our diagnosis. IPFDs are thus rare heritable bleeding diseases in children but are also one of the most difficult diagnostic challenges, even for experienced clinicians/laboratories. A large heterogeneity in the clinical presentation and diagnostic approaches represent serious limitations to the identification of these rare disorders. Diagnosis is important for appropriate, timely management and to prevent the occurrence of life-threatening bleeding manifestations and severe anemia. Prevention of bleeding is the best approach for these patients. Extensive awareness and education programs must be conducted for families affected by these rare disorders.
CONCLUSION
This case series emphasizes the rare and varied presentations of GT and BSS. A high index of suspicion must be considered in pediatric patients presenting with bleeding manifestations so as not to delay the timely diagnosis and treatment. Awareness and utilizing BS, along with family studies and collaborative multidiagnostic modality, is important to reach a conclusive diagnosis in these rare disorders.
Acknowledgments
The authors would like to thank Dr. Punnet Kaur Sahi, Dr. Shelly Mittal, and Dr. Deepshikha, Department of Pediatrics helped in the clinical diagnosis and management of the patient.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Inherited platelet-based bleeding disorders. J Thromb Haemost. 2003;1:1628-36.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited platelet disorders. Hematology Am Soc Hematol Educ Program 2005:396-403. doi: 10.1182/asheducation-2005.1.396
- [CrossRef] [PubMed] [Google Scholar]
- Congenital platelet disorders and understanding of platelet function. Br J Haematol. 2014;165:165-78.
- [CrossRef] [PubMed] [Google Scholar]
- Management of siblings with Glanzmann's thrombasthenia: A case report. J Family Med Prim Care. 2020;9:1733-5.
- [CrossRef] [PubMed] [Google Scholar]
- A rare case report on Glanzmann thrombasthenia. Natl J Physiol Pharm Pharmacol. 2017;7:1291-2.
- [CrossRef] [Google Scholar]
- Glanzmann thrombasthenia and Bernard-Soulier syndrome in south Iran. Clin Lab Haematol. 2005;27:324-7.
- [CrossRef] [PubMed] [Google Scholar]
- Glanzmann thrombasthenia: State of the art and future directions. Semin Thromb Hemost. 2013;39:642-55.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited platelet functional disorders: General principles and practical aspects of management. Transfus Apher Sci. 2018;57:494-501.
- [CrossRef] [PubMed] [Google Scholar]
- Profiling the genetic and molecular characteristics of Glanzmann Thrombasthenia: Can it guide current and future therapies? J Blood Med. 2021;12:581-99.
- [CrossRef] [PubMed] [Google Scholar]
- Bernard-Soulier syndrome (hemorrhagic parous throm-bocytic dystrophy) Orphanet J Rare Dis. 2006;1:46.
- [CrossRef] [PubMed] [Google Scholar]
- Functional and molecular characterization of inherited platelet disorders in the Iberian Peninsula: Results from a collaborative study. Orphanet J Rare Dis. 2014;9:213.
- [CrossRef] [PubMed] [Google Scholar]




