
Translate this page into:
Multicentric Castleman disease involving the bone marrow in an immunocompetent patient masquerading as a non-Hodgkin lymphoma – A diagnostic conundrum

*Corresponding author: Soundarya Ravi, Department of Pathology, Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India. amsosmiles@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Ravi S, Balasubramaniam V, Kannan N, Manivannan P, Kar R. Multicentric Castleman disease involving the bone marrow in an immunocompetent patient masquerading as a non-Hodgkin lymphoma – A diagnostic conundrum. J Hematol Allied Sci. 2025;5:103-8. doi: 10.25259/JHAS_21_2024
Abstract
Multicentric Castleman disease (MCD) is a polyclonal hematological disorder characterized by constitutional symptoms, organomegaly, significant lymphadenopathy, cytopenias, and symptoms of hypergammaglobulinemia, attributed to excessive interleukin-6 production. The clinical presentation, as well as the bone marrow features of MCD, is not unique and overlaps with a myriad of diseases such as chronic infections, autoimmune diseases, and hematological neoplasms, creating diagnostic difficulty. In this case report, we describe the bone marrow pathology of MCD diagnosed in an elderly immunocompetent patient who presented with clinical features of non-Hodgkin lymphoma, along with a brief discussion on its differential diagnosis.
Keywords
Bone marrow
Castleman disease
Flow cytometry
Multicentric
Non-Hodgkin lymphoma
Plasma cell dyscrasia

INTRODUCTION
Castleman disease (CD) is a unique hematological disorder that is characterized by diverse clinical presentation and histopathological features. According to the 5th edition of the World Health Organization (WHO) Classification of Hematolymphoid Tumors, CD is placed under a specific category of “tumor-like lesions with B-cell predominance.”[1] The most common histopathological pattern observed in the lymph node of CD patients is hyaline vascular (HV) variant followed by the plasmacytic variant (PC) and mixed variant displaying features intermediate between HV and PC variants.[2,3]
At present, CD is classified based on the clinical presentation into unicentric CD when there is localized involvement of lymph nodes and multicentric CD (MCD) when there is multifocal lymphadenopathy accompanied by systemic involvement. According to the etiopathogenesis, MCD is further subclassified into three subtypes: Human herpes virus-8-associated MCD (HHV-8-associated MCD), polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes (POEMS)-associated MCD, and idiopathic MCD (iMCD).[4] Furthermore, iMCD is subdivided into two types, namely iMCD associated with thrombocytopenia, anasarca, fever, reticulin myelofibrosis and renal dysfunction and organomegaly (iMCD-TAFRO), and iMCD not otherwise specified.[5] HHV-8-associated MCD is commonly noted among immunocompromized individuals and people living with human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome. iMCD is a diagnosis of exclusion made after the elimination of all possible etiological causes of MCD.[6]
Regardless of the subtype, MCD can present with multifocal lymphadenopathy, organomegaly, profound systemic inflammatory symptoms, multiorgan dysfunction, cytopenias, and polyclonal hypergammaglobulinemia. These manifestations are attributed to excessive cytokine production, especially interleukin-6 (IL-6).[7] Nevertheless, these clinical manifestations and associated laboratory findings are not unique to MCD and can also overlap with a variety of other disorders such as chronic infections, auto-immune diseases, and hematological neoplasms such as Hodgkin lymphoma (HL), non-HL (NHLs), and plasma cell dyscrasias (PCD).[8] Despite the recent development of integrated diagnostic criteria for the diagnosis of MCD, the diagnosis remains challenging.[6]
A lymph node biopsy in the first place would aid in clinching the diagnosis of CD. However, in situations where the primary diagnostic material obtained by the pathologist is bone marrow aspiration and biopsy, it is essential to be familiar with the histopathological findings of MCD in the marrow, to rule out its morphological mimics. In this case report, we describe bone marrow involvement by MCD in an immunocompetent patient presenting with clinical features of NHL.
CASE REPORT
A 70-year-old gentleman presented to the Medicine outpatient department with a 2-month history of low-grade, intermittent fever, fatigue, giddiness, shortness of breath, and weight loss. In addition, he also reported having few episodes of bleeding gums for the past week. On examination, he had pallor, moderate splenomegaly, and hepatomegaly extending up to 2 cm below the right costal margin. He also had bilateral non-tender cervical and axillary lymphadenopathy, with the largest lymph node measuring 1 cm located in the left cervical region. His central nervous system and peripheral nervous system examination revealed no significant abnormalities.
Complete hemogram showed anemia and thrombocytopenia, with an erythrocyte sedimentation rate of 160 mm at the end of 1st h. Peripheral smear examination revealed red blood cells demonstrating rouleaux formation with 4% lymphoplasmacytoid cells [Figure 1a]. Biochemical analysis indicated elevated total protein levels with reversal of albumin/globulin ratio. The results of the complete hemogram and biochemical investigations of this patient are summarized in Table 1. Radiological examination was performed which did not show any bone lytic lesions. Based on the clinical presentation and the results of the basic biochemical and hematological investigations performed, there was a strong clinical suspicion of NHL, particularly lymphoplasmacytic lymphoma followed by PCD and our patient was further worked up in this regard.
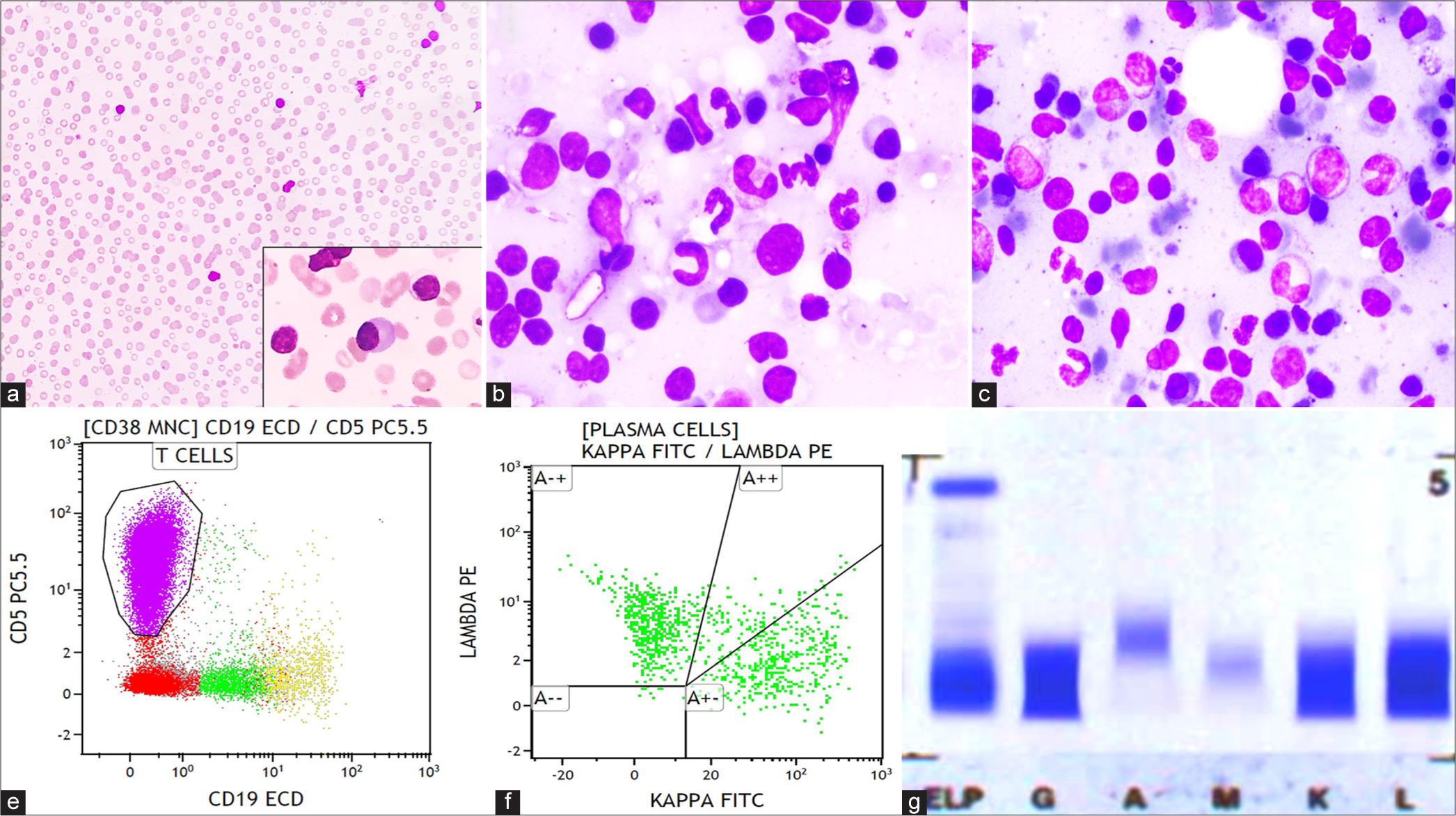
- (a) Peripheral smear shows rouleaux formation of red blood cells (×40; Giemsa) with occasional plasmacytoid lymphocytes (within inset ×400; Giemsa). (b and c) Bone marrow aspirate smear shows lymphocytes, plasma cells, and lympho-plasmacytoid cells admixed with hematopoietic cells (×400; Giemsa). (d) Flow cytometry (FCM) of the bone marrow aspirate shows that the lymphocyte population is primarily composed of T-cells followed by B-cells. (e) FCM also shows that plasma cells are polyclonal for kappa and lambda. (f) Immunofixation shows positive bands in immunoglobulin G, kappa, and lambda.
| Test name | Patient’s test results | Biological reference interval |
|---|---|---|
| Hemoglobin level | 69 g/L | 140–160 g/L |
| Total leukocyte count | 10.3×109/L | 4–11×109/L |
| Platelet count | 30×109/L | 150–450×109/L |
| Total protein | 11.17 g/dL | 5.5–9.0 g/dL |
| Serum albumin | 1.55 g/dL | 3.5–5.5 g/dL |
| Serum globulin | 9.62 g/dL | 2.0–3.5 g/dL |
| Serum creatinine | 0.8 mg/dL | 0.7–1.3 mg/dL |
| Serum calcium | 8.2 mg/dL | 8.5–10.5 mg/dL |
| Lactate dehydrogenase | 277 U/L | 125–220 U/L |
| Serum β2 microglobulin | 12,775.84 ng/mL | 800–2,340 ng/mL |
Subsequently, bone marrow aspiration and trephine biopsy were performed. Bone marrow aspirate smears were cellular and showed prominence of small-sized lymphoid cells and mature plasma cells admixed with lympho-plasmacytoid cells [Figure 1b and c]. Erythropoiesis and myelopoiesis were suppressed with a few preserved megakaryocytes. No hemophagocytosis or organisms were observed. Bone marrow biopsy was hypercellular and showed diffuse marrow fibrosis (WHO grade 2 reticulin fibrosis). There were multiple large nodular aggregates composed of a polymorphous population of lymphoid cells surrounded by a rim of mature appearing plasma cells in the intertrabecular spaces. At places, these plasma cells were arranged in sheets and formed large aggregates. The interstitium also showed many singly scattered lymphoid cells with increased vascular proliferation. In addition, there was megakaryocytic hyperplasia forming loose clusters and exhibiting atypical features in the form of hypolobated nucleus and separation of lobes. On immunohistochemistry, these lymphoid nodules were composed predominantly of T-cells admixed with few B-cells, as highlighted by CD3 and CD20, respectively. The rim of plasma cells surrounding the lymphoid nodules was highlighted by CD138 [Figure 2]. The presence of T-cell-rich nodules in the marrow raised a suspicion of bone marrow infiltration by T-cell NHL associated with plasmacytosis and secondary para-proteinemia.
![(a) Bone marrow biopsy shows hypercellular marrow with large nodular aggregates composed of small lymphoid cells surrounded by a rim of plasma cells (×40; Hematoxylin and Eosin [H&E]). (b) Bone marrow biopsy shows megakaryocytic hyperplasia forming loose clusters with few monolobated forms (×400; H & E). (c) Reticulin stain highlights diffuse World Health Organization grade 2 fibrosis (×40). (d and e) CD3 and CD20 highlights the lymphoid aggregates in the marrow (×40). (f) CD138 highlights the plasma cells aggregates in the interstitium forming a rim around the lymphoid nodules (×40).](/content/129/2025/5/1/img/JHAS-5-103-g002.png)
- (a) Bone marrow biopsy shows hypercellular marrow with large nodular aggregates composed of small lymphoid cells surrounded by a rim of plasma cells (×40; Hematoxylin and Eosin [H&E]). (b) Bone marrow biopsy shows megakaryocytic hyperplasia forming loose clusters with few monolobated forms (×400; H & E). (c) Reticulin stain highlights diffuse World Health Organization grade 2 fibrosis (×40). (d and e) CD3 and CD20 highlights the lymphoid aggregates in the marrow (×40). (f) CD138 highlights the plasma cells aggregates in the interstitium forming a rim around the lymphoid nodules (×40).
Immunophenotyping by flow cytometry (FCM) of the bone marrow aspirate was performed using three tubes-10 color panel, as detailed in Table 2. FCM revealed that the lymphocyte population was primarily composed of T-cells (75.5%), followed by polyclonal B-cells (2.9%) [Figure 1d]. Among the T-cells, 92.7% expressed TCR alpha-beta, and 5% expressed TCR gamma-delta, with a CD4:CD8 ratio of 1:1.87. The T-cells did not express any aberrant markers, nor was there any downregulation of T-cell markers. Plasma cells accounted for only 1% of the population, possibly due to a dilution effect, and were polyclonal in nature with a kappa: lambda ratio of 1:1.4, as detected by cytoplasmic staining using light chain antibodies [Figure 1e]. Both plasma cells and B-cells were negative for surface immunoglobulin M (IgM). There was no evidence of an abnormal T-cell population or clonal populations in B-cells and plasma cells, effectively ruling out the possibility of both NHL and PCD.
| Tube | FITC | PE | ECD | PC5.5 | PC7 | APC | APC 700 | APC 750 | PB | KO |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 (Clone) | – | – | CD19 | CD5 | CD200 | CD23 | CD20 | CD43 | CD22 | CD45 |
| – | – | J3-119 | BL1a | OX-104 | 9P25 | HRC20 | DFT1 | SJ10.1H11 | J33 | |
| 2 (Clone) | Cyto-Kappa | Cyto-Lambda | CD117 | CD19 | CD56 | CD138 | CD28 | CD38 | – | CD45 |
| Polyclonal F (ab’) 2 Goat | Polyclonal F (ab’) 2 Goat | 104D2D1 | J3-119 | N901 | B-A38 | LS198-4-3 | – | J33 | ||
| 3 (Clone) | CD8 | CD26 | CD45 | TCR-γδ | CD5 | TCR-αβ | CD4 | CD7 | sCD3 | CD16/CD56 |
| B9.11 | 4EL-1C7 | J33 | IMMU510 | BL1a | IP26A | 13B8.2 | 8H8.1 | UCHT1 | 3G8 |
Serum protein electrophoresis (SPEP) showed diffuse band in the gamma globulin region with no detection of M-band. Immunofixation electrophoresis (IFE) revealed polyclonal hypergammaglobulinemia which was positive for immunoglobulin G (IgG), kappa, and lambda immunoglobulins [Figure 1f]. Serum immunoglobulin levels were measured which revealed elevated IgG and IgM levels. Serum free light chain (SFLC) assay revealed elevated levels of both free kappa and lambda light chains with kappa: lambda ratio of 0.43. The results of the serum immunoglobulin levels and SFLC assay of this patient are summarized in Table 3. At this point, additional investigations were conducted to rule out infectious and auto-immune conditions causing polyclonal hypergammaglobulinemia. Serology for HIV, hepatitis B virus, hepatitis C virus, and Epstein–Barr virus along with tuberculosis and auto-immune workup yielded negative results. The parameters included in the diagnostic criteria for hemophagocytic lymphohistiocytosis, such as serum ferritin, triglycerides, liver enzymes, and fibrinogen, were within normal ranges.
| Test name | Serum levels | Biological reference interval |
|---|---|---|
| Serum IgG level | 71.67 g/L | 6.10–16.16 g/L |
| Serum IgA level | 4.07 g/L | 0.84–4.99 g/L |
| Serum IgM level | 4.16 g/L | 0.35–2.42 g/L |
| Serum-free kappa light chain | 380.94 mg/L | 3.3–19.4 mg/L |
| Serum-free lambda light chain | 876.56 mg/L | 5.71–26.30 mg/L |
| Free kappa: lambda ratio | 0.43 | 0.26–1.65 |
IgG: Immunoglobulin G, IgA: Immunoglobulin A, IgM: Immunoglobulin M
Eventually, left cervical lymph node biopsy was performed. The histopathological and immunohistochemical features of the lymph node biopsy were consistent with plasma cell variant of CD [Figure 3]. The lymph node morphology threw light upon our patient’s diagnosis which was finally identified as MCD. However, due to the unavailability of HHV-8 LANA-1 immunostain, we were unable to rule out the possibility of HHV-8-associated MCD. Unfortunately, due to unforeseen circumstances, the patient succumbed to the illness before the initiation of chemotherapy and further investigations, such as serum IL-6 and vascular endothelial growth factor level assessments, could be performed.
![(a and b) Lymph node biopsy shows paracortical expansion by sheets of plasma cells with prominent vasculature accompanied by regressed follicles (a– ×40; Hematoxylin and Eosin [H&E], b– ×100; H&E). (c) CD23 highlights the dendritic cells in the regressed follicles (×100). (d) CD38 highlights the plasma cells in the paracortical region (×100).](/content/129/2025/5/1/img/JHAS-5-103-g003.png)
- (a and b) Lymph node biopsy shows paracortical expansion by sheets of plasma cells with prominent vasculature accompanied by regressed follicles (a– ×40; Hematoxylin and Eosin [H&E], b– ×100; H&E). (c) CD23 highlights the dendritic cells in the regressed follicles (×100). (d) CD38 highlights the plasma cells in the paracortical region (×100).
DISCUSSION
Numerous studies have extensively described the histopathological features of lymph nodes in MCD. However, only a limited number of studies in the literature focus on the bone marrow findings in patients with MCD.[9-12] The bone marrow in these patients typically exhibits high cellularity marrow plasmacytosis which is a prominent feature of MCD, documented in both HHV-8-associated MCD and iMCD. Increased IL-6 production stimulates plasma cell proliferation, leading to the formation of large interstitial aggregates resembling plasma cell myeloma (PCM). Unlike PCM, the plasma cells in MCD are reactive, exhibit polyclonality, and display a mature morphology, as observed in our patient.[9,11] Nevertheless, some studies have reported the presence of immature plasma cells and plasmablasts with light chain restriction, especially in HHV-8-associated MCD.[10] Various inflammatory conditions, including autoimmune disorders, chronic infections such as syphilis and leishmaniasis, and liver disease, can also exhibit reactive marrow plasmacytosis, posing a close differential diagnosis for MCD.[13] However, in these conditions, the reactive plasma cells are typically scattered or form small clusters, which are generally located perivascularly.
A few studies in the literature describe the presence of multiple lymphoid aggregates in the bone marrow biopsy of MCD with a higher frequency noted in HHV-8-associated MCD when compared to iMCD.[9] These lymphoid aggregates are composed of a mixture of T-cells and B-cells. Ibrahim et al. reported B-cell-predominant lymphoid aggregates in one of their cases of MCD.[10] In our case, there was a predominance of T-cells in the lymphoid aggregates, prompting the consideration of a T-cell NHL in our differential diagnosis. Moreover, these lymphoid nodules were surrounded by a rim of plasma cells, a pattern typically described in the bone marrow of patients with POEMS syndrome.[14] However, the differential diagnosis of NHL was ruled out by FCM analysis of the bone marrow, which did not detect any clonal B-cell populations or any aberrant phenotypes in T-cells. The polyclonal nature of plasma cells detected in FCM and the presence of polyclonal hypergammaglobulinemia identified through SPEP and IFE helped rule out PCM and POEMS syndrome.
Additional findings noted in our patient’s marrow included hyperplasia of megakaryocytes forming loose clusters associated with megakaryocytic atypia, along with the presence of diffuse WHO grade 2 reticulin fibrosis. Atypical features noted in the megakaryocytes were the presence of hypolobated and hyperchromatic forms. Megakaryocytic hyperplasia, megakaryocytic atypia, and higher grades of reticulin fibrosis were commonly reported in iMCD patients, especially in a recently introduced subtype known as iMCD-TAFRO group. However, these findings are not very specific to MCD and can be seen even in many other inflammatory and neoplastic processes. Belyaeva et al. postulated that megakaryocytic hyperplasia represented the marrow’s response to immune-mediated platelet destruction.[9] It was also believed that increased levels of inflammatory cytokines were responsible for causing marrow fibrosis and megakaryocytic atypia. Some studies have also described hemophagocytosis in the marrow of MCD patients.[10] However, there was no evidence of hemophagocytosis in our case.
CONCLUSION
Even though the bone marrow findings of MCD have been described in the literature, these findings are not very specific to MCD and can overlap with a variety of inflammatory, infective, and neoplastic conditions. Therefore, the diagnosis of MCD in the bone marrow is challenging and necessitates the integration of clinical, hematological, immunological, and histological workup to provide an accurate and prompt diagnosis.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of AI-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- The 5th edition of the World Health Organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia. 2022;36:1720-48.
- [CrossRef] [PubMed] [Google Scholar]
- Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29:670-83.
- [CrossRef] [PubMed] [Google Scholar]
- Castleman disease: An update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009;16:236-46.
- [CrossRef] [PubMed] [Google Scholar]
- Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol. 2019;94:975-83.
- [CrossRef] [PubMed] [Google Scholar]
- International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129:1646-57.
- [CrossRef] [PubMed] [Google Scholar]
- Brief report: Alleviation of systemic manifestations of Castleman's disease by monoclonal anti-interleukin-6 antibody. N Engl J Med. 1994;330:602-5.
- [CrossRef] [PubMed] [Google Scholar]
- Idiopathic multicentric Castleman's disease: A systematic literature review. Lancet Haematol. 2016;3:e163-75.
- [CrossRef] [PubMed] [Google Scholar]
- Bone marrow findings of idiopathic Multicentric Castleman disease: A histopathologic analysis and systematic literature review. Hematol Oncol. 2022;40:191-201.
- [CrossRef] [PubMed] [Google Scholar]
- Bone marrow manifestations in multicentric Castleman disease. Br J Haematol. 2016;172:923-9.
- [CrossRef] [PubMed] [Google Scholar]
- Bone marrow findings in multicentric Castleman disease in HIV-negative patients. Am J Surg Pathol. 2007;31:398.
- [CrossRef] [PubMed] [Google Scholar]
- Bone marrow involvement in Multicentric Castleman disease in a HIV negative patient. Indian J Hematol Blood Transfus. 2014;30:60-3.
- [CrossRef] [PubMed] [Google Scholar]
- Bone marrow plasmacytosis in idiopathic plasmacytic lymphadenopathy with polyclonal hyperimmunoglobulinemia: A report of four cases. Pathol Res Pract. 2007;203:789-94.
- [CrossRef] [PubMed] [Google Scholar]
- Bone marrow histopathology in POEMS syndrome: A distinctive combination of plasma cell, lymphoid, and myeloid findings in 87 patients. Blood. 2011;117:6438-44.
- [CrossRef] [PubMed] [Google Scholar]




