
Translate this page into:
Genotypic and hematologic association of pulmonary hypertension in non-transfusion-dependent thalassemia
*Corresponding author: Prakas Kumar Mandal, Department of Clinical Haematology, Nil Ratan Sircar Medical College and Hospital, Kolkata, West Bengal, India. prakas70@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Vinodhini M, De R, Mandal PK, Dutta S. Genotypic and hematologic association of pulmonary hypertension in non-transfusion-dependent thalassemia. J Hematol Allied Sci. doi: 10.25259/JHAS_11_2025
Abstract
Objectives
Thalassemia intermedia phenotype, older age, splenectomy, hypercoagulability, chronic hemolysis, hypoxia, nitric oxide depletion, endothelial perturbation, and iron overload contribute to pulmonary hypertension in thalassemia. We aimed to explore the association of genotype and disease parameters on pulmonary hypertension in non-transfusion-dependent thalassemia (NTDT).
Material and Methods
Prospective cross-sectional data comprising demographics, disease characteristics, genotype, and pulmonary hypertension on 50 non-transfusion-dependent hemoglobin (Hb) E beta-thalassemia patients were collected.
Results
The majority (n = 32,64%) required either one or no transfusion and had median Hb of 8.5 g/dL (range 5.9–14.1) and median fetal hemoglobin (Hb F) of 36.8% (range 2.5–76.8%). A mild Mahidol score was seen in 42 (84%) cases. Pulmonary hypertension was detected in 32%, grade I disease in 28%, and none were symptomatic. High Hb F was noted where compound heterozygosity for intervening sequence (IVS)1-5(G>C)+codon(CD)26(G>A) beta-globin mutation and alpha(-α)3.7 and -α4.2 deletion were detected. Mean pulmonary artery pressure (MPAP) was high when Hb F >25% but was not significant (P = 0.67). The odds ratio for pulmonary hypertension at serum ferritin more than 500 ng/ml was 9.4 (confidence interval 2.58-33.27, P = 0.00038). Older patients had high ferritin (P = 0.0025). Around 25% variability in MPAP was influenced by age, number of transfusions, ferritin, Hb F, and Hb. None underwent splenectomy or had prior thromboembolism or heart failure.
Conclusion
In spite of young age, mild Mahidol score, and high Hb F in those with alpha deletion and beta mutation, pulmonary hypertension was still a significant complication even in the absence of splenectomy or prior thromboembolic episode in non-transfusion-dependent thalassemia. As this was the foremost study investigating the relationship of genotypes with complications in NTDT in India, future multicenter studies are paramount.
Keywords
Alpha globin gene deletion
Beta globin gene mutation
Hemoglobin E beta thalassemia
Nontransfusion-dependent thalassemia
Pulmonary hypertension

INTRODUCTION
Approximately 55,000 children are diagnosed with severe forms of thalassemia, of which 20% carry single gene deletion.[1] In non-transfusion-dependent thalassemia (NTDT), transfusion is essential only during increased demand, such as pregnancy, infection, and growth failure. Primary disease modifiers are beta gene mutation resulting in variable reduction (β+) to complete absence of β-globin (β0), and secondary modifiers are coinheritance of mutations in α- and γ-chains, γ-chain expression, polymorphisms in Xmn1, BCL11A, KLF1, and HBS1LMYB genes influencing hemoglobin (Hb) F.[2-4] No association was noted between genotype and complications in homozygous beta-thalassemia.[5,6] The objective was to analyze the association between pulmonary hypertension and disease parameters in NTDT.
MATERIAL AND METHODS
This prospective open-label cross-sectional observational study comprised 50 non-transfusion-dependent (NTD) hemoglobin (Hb) E beta-thalassemia patients who attended a thalassemia clinic in a tertiary care hospital. Patients were followed up every 3 months. Patients aged 5 years and above diagnosed with NTD Hb E beta-thalassemia and without chelation therapy were included as study participants. Patients with co-morbidities such as chronic obstructive or restrictive pulmonary diseases and congenital or acquired valvular or pericardial heart diseases were excluded from the study. By the 1964 Helsinki Declaration and its later amendments, as well as by the ethical standards of the institutional and national research committee, human participants were subjected to all procedures. The study participants were recruited after approval by the institute ethics committee (approval number: NMC/000182). Informed consent (or assent when applicable) was obtained from all study participants in the analysis.
Study parameters
A detailed clinical history, examination and evaluation for inter-current infections, leg ulcers, thrombo-embolic complications, endocrine disturbances such as diabetes mellitus, hypothyroidism, growth retardation, hypogonadism, menstrual history, extra-medullary hematopoiesis, chelation therapy, and splenectomy were undertaken. Complete hemogram, liver and renal function test, viral marker for hepatitis B, hepatitis C and human immunodeficiency virus, random blood glucose, liver stiffness measurement by transient elastography, serum ferritin by chemiluminescence assay, Mahidol score incorporating age during the first presentation to the clinic, age during first blood transfusion, number of blood transfusion received, hemoglobin level, size of spleen and growth as well as development were assessed. Mild disease was defined as a score of zero to four, moderate as a score of four to seven, and a score of 7–10 as severe disease. Cation exchange-high performance liquid chromatography (BioRad Laboratories, California, USA) analyzed the thalassemia screening investigation using the Variant II β-thal short program. Pulmonary hypertension was defined as an increase in mean pulmonary artery pressure (MPAP) of more than 25 mmHg or tricuspid regurgitation velocity (TRV) of more than 2.7 m/sec by conventional Doppler echocardiography. Alpha globin gene deletion analysis was done using gap polymerase chain reaction (PCR), and beta-globin gene mutation was tested using amplification refractory mutation system PCR. Skiagrams of the chest, long bones, ultrasonography, bone mineral densitometry, thyroid function test, and coagulation screen were carried out when clinically indicated. Hydroxyurea was administered to all patients. Chelation was initiated when ferritin was more than 800 ng/mL or as per clinical indication when complications were developed.
Statistical analysis
Data were analyzed using the Statistical Package for the Social Sciences version 21.0. Categorical variables were represented as frequencies and percentages, and numerical variables were reported as mean ± standard deviation or median and interquartile range. The difference between groups regarding categorical variables was evaluated using the Chi-square test, Analysis of Variance (ANOVA), Kruskal–Wallis test, and odds ratio (OR). For statistical significance, a P < 0.05 was considered. An independent sample t-test or Mann–Whitney U-test was used to test the difference in two mean values. Correlation was evaluated by correlation and regression analysis.
RESULTS
We evaluated 50 NTD Hb E beta-thalassemia patients. The median age was 21 years (range 5–45 years). There were 27 males (54%) and 23 females (46%). The demographic, disease, and clinical characteristics are shown in Table 1. Clinically, pallor, mild icterus, and thalassemic facies were present. Most were either never transfused or received occasional transfusions. We found pulmonary hypertension in 16 cases (32%). None were symptomatic. About 14 (28%) had grade I, and two (4%) had moderate disease (grade II). The majority, n = 32(64%), required no transfusion or only one transfusion, had a median Hb of 8.5 g/dl (5.9–14.1) and had a median fetal hemoglobin (Hb F) level of 36.8% (2.5– 76.8%). People receiving more transfusion units (> three) had low Hb F, high Hb A2/E, and high ferritin. Thirteen patients (26%) had ferritin more than 500 ng/mL and three (6%) had more than 800 ng/mL and required chelation therapy. Around 42 (84%) had mild Mahidol scores, of which 30 (60%) had ferritin <500 ng/mL. Eight (16%) had a moderate Mahidol score. There were no thrombosis, hypogonadism, diabetes mellitus, pseudoxanthoma elasticum, leg ulcers, osteoporosis, portal vein thrombosis, viral hepatitis, or extra-medullary pseudo-tumors. None underwent splenectomy.
| Disease and genotype characteristics | Number of patients, n=50 (%) |
|---|---|
| Age <25 years | 36 (72) |
| Male: female ratio | 1.17 |
| Mild Mahidol score | 42 (84) |
| Presence of pulmonary hypertension | 16 (32) |
| Presence of beta-globin gene mutation (out of 31 cases tested) | 23 (46) |
| Presence of alpha globin gene deletion (out of 27 cases tested) | 14 (28) |
| Presence of hypothyroidism (out of 18 cases tested) | 3 (16) |
| Presence of hepatic fibrosis | 1 (2) |
| Disease parameters | Median (IQR) |
| Number of transfusions in each patient | 1 (0–6) |
| Hemoglobin in g/dL | 8.5 (5.9–14.1) |
| Fetal hemoglobin level in percentage | 36.8 (2.5–76.8) |
| Serum ferritin in ng/mL | 421 (91.4–1098) |
| Total bilirubin in mg/dL | 3.9 (0.7–9.6) |
| Indirect bilirubin in mg/dL | 3.5 (0.5–9.3) |
| Blood urea in mg/dL | 27 (14–48) |
| Serum creatinine in mg/dL | 0.8 (0.5–1.5) |
| Random blood glucose in mg/dL | 95 (63–120) |
IQR: Interquartile range
Genotype analysis
Beta globin gene mutation testing was done in 31/50(62%). Compound heterozygosity for intervening sequence (IVS)1-5(G>C)+ codon (CD)26(G>A) beta-globin gene mutation was detected in 19 (38%), CD30(G>C)+CD26(G>A) beta-globin gene mutation in four (8%), CD15(G>A)+CD26(G>A), CD8/9(+G)+CD26(G>A), CD41/42(-CTTT)+CD26(G>A), and IVS 1-5(G>C) homozygous beta-globin gene mutations were seen in two patients (4%) each. A higher proportion of IVS1-5 mutations had a Hb F level of more than 25% compared to other beta gene mutations, as shown in Figure 1.

- Fetal hemoglobin level in various beta globin gene mutation. Hb F: Hemoglobin F.
Alpha globin gene deletions -α3.7 and -α4.2 deletion were done in 27/50 (54%). Out of a total of 19 who showed IVS 1-5(G>C)+CD26(G>A)beta gene mutation, 13 showed alpha gene deletion, four were normal, and an alpha gene mutation study was not done in two cases. Out of 13 with IVS 1-5(G>C)+CD26(G>A)beta mutation with alpha gene deletion, αα/α-3.7 was the most common and was detected in six cases. α-4.2/α-4.2 was detected in four and αα/α-4.2 was noted in three cases. Out of four with CD30(G>C)+CD26(G>A) beta-globin gene mutation, three showed alpha globin gene deletion mutation, and in one, alpha globin gene deletion, analysis was not done. Out of three with CD30(G>C)+CD26(G>A) beta-globin gene mutation with alpha-globin gene deletion, αα/α-3.7 was detected in two and α-4.2/α-4.2 in only one case. Only one with CD 41/42 beta-globin gene mutation showed α-4.2/α-4.2 globin gene deletion. When alpha gene deletions were present, Hb F levels were higher than when alpha deletions were absent, as described in Figure 2.
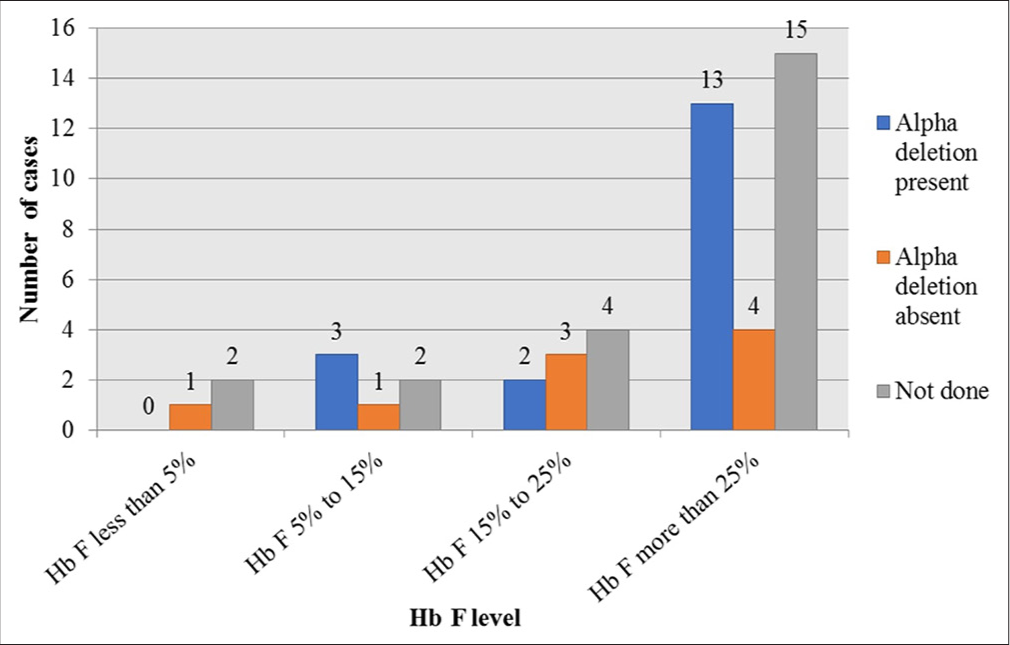
- Fetal hemoglobin level depending upon alpha globin gene deletion. Hb F: Hemoglobin F.
A high proportion of IVS 1-5 beta mutation and alpha deletion had high Hb F compared to other beta-globin gene mutations or when alpha gene deletion was absent. This finding may have resulted from the preferential affinity of residual alpha globin chains with gamma globin chains compared to mutant or normal beta-globin gene polypeptide chains.
Analysis of hematologic parameters
Pulmonary hypertension and Mahidol score: Early age at first presentation and low average Hb level contributed to a high Mahidol score and high incidence of pulmonary hypertension. A clinical and statistically significant association was noted between pulmonary hypertension, high Mahidol score (P = 0.030), and low left ventricular ejection fraction (P = 0.035). There was no significant difference in other hematologic and disease parameters, as depicted in Table 2.
| Disease characteristics | Total (n=50) | Pulmonary hypertension | P-value | |
|---|---|---|---|---|
| Absent (n=34) | Present (n=16) | |||
| Mahidol score | 4.0 (1.0–7) | 2.0 (1.0–7) | 4.0 (1.0–7) | 0.030 |
| Left ventricular ejection fraction in percentage | 65.5 (61.0–69.0) | 67.0 (64.0–68.0) | 62.0 (61.0–69.0) | 0.035 |
| Liver size below the costal margin, in centimeters | 3.0 (3.0–6.0) | 3.0 (3.0–5.0) | 4.0 (3.0–6.0) | 0.36 |
| Spleen size below the costal margin, in centimeters | 4.1±1.7 | 4.1±1.5 | 4.1±1.7 | 0.94 |
| Hemoglobin F, in percentage | 35.5±20.8 | 36.5±20.4 | 33.5±17.9 | 0.63 |
| Hemoglobin A0, in percentage | 6.8 (5.0–10.0) | 7.3 (5.2–10.0) | 6.7 (5.0–9.1) | 0.35 |
| Hemoglobin A2/E, in percentage | 55.2±19.9 | 55.2±19.8 | 56.7±16.9 | 0.79 |
| Hemoglobin in g/dL | 8.5 (5.9–14.1) | 7.5 (5.9–12.2) | 7.8 (6.4–14.1) | 0.93 |
| Red blood cell count in million/cubic mm | 4.1 (3.4–5.1) | 4.0 (3.5–4.3) | 4.3 (3.4–5.1) | 0.48 |
| Red cell distribution width, in percentage | 23.5±5.7 | 23.6±5.6 | 23.4±3.2 | 0.89 |
| Serum ferritin, ng/mL | 421 (91.4–1098) | 311 (91.4–433.0) | 412 (251.2–1098) | 0.11 |
| Total bilirubin, mg/dL | 3.9 (3.2–4.9) | 3.9 (3.2–4.6) | 4.0 (3.2–4.9) | 0.77 |
For normally distributed variables, descriptive statistics were represented as mean±standard deviation, and for non-normally distributed parameters, median (interquartile range) was defined. For statistical significance, the P-value was set at<0.05, Bold denotes Significant P value.
Pulmonary hypertension and age: When the grades of pulmonary hypertension were considered about age, no significant difference in the grade of pulmonary hypertension between age groups <25 years and >25 years was identified in the study (Chi-square=0.33, df = 1 P = 0.5).
Pulmonary hypertension and Hb: When Hb level cutoff of 7 g/dL was used to divide the cases into two groups, no significant difference in the incidence of pulmonary hypertension was detected between the groups <7 g/dL and more than 7 g/dL (Chi-square 0.02, df = 1, P = 0.887) although there was a weak positive correlation of MPAP with Hb level (r = 0.17, r2 = 0.02).
Pulmonary hypertension and Hb F level: On analysis of the MPAP distribution in three groups defined by Hb F cut-offs of <15%, 15–25%, and more than 25%, the median MPAP tended to be higher in Hb F more than 25% subgroup compared to other two groups. However, the difference obtained was not significant (ANOVA SS = 41.12, df = 2, MS = 20.56, F = 0.4, P = 0.67); it was observed that the median MPAP was higher in the group with the highest Hb F level as shown in Figure 3.
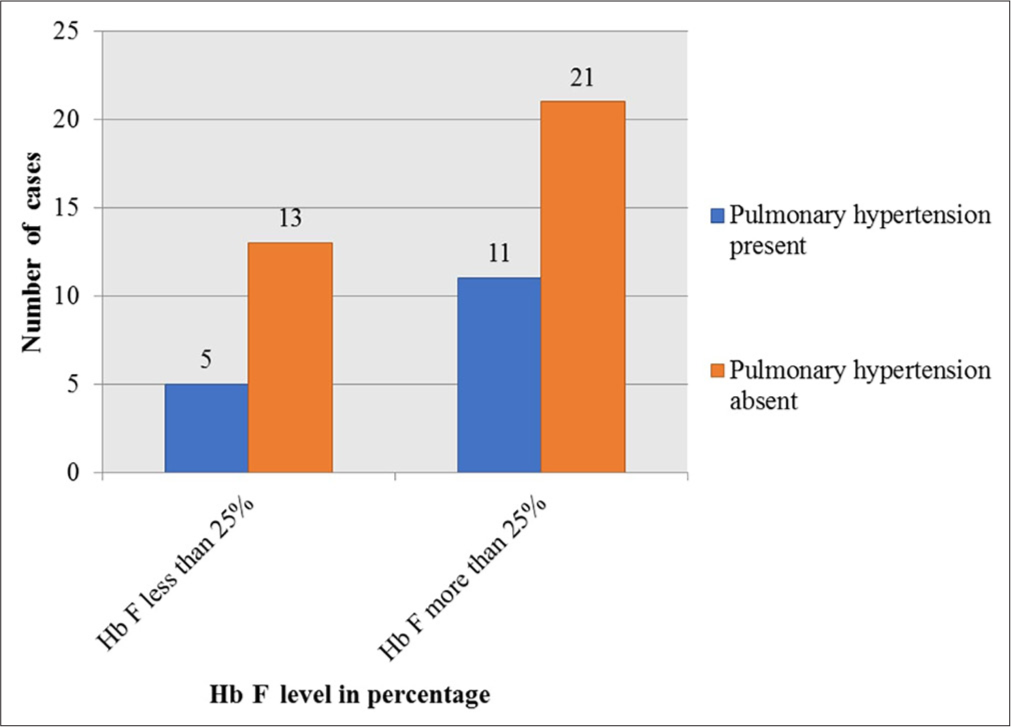
- Association of pulmonary hypertension and fetal hemoglobin level. Hb F: Hemoglobin F.
Pulmonary hypertension and serum ferritin: There was an association between serum ferritin and the presence of pulmonary hypertension, as shown in Figure 4. When a cut-off level of ferritin 500 ng/mL was considered, there was a higher proportion of patients having pulmonary hypertension than when ferritin was >500 ng/mL (Chi-square test = 0.33; df = 1; P = 0.021). The OR for MPAP ≥25 mm Hg at serum ferritin more than 500 ng/mL was 9.4 (confidence interval [CI] 2.58–33.27, P = 0.00038). Furthermore, serum ferritin had a weak positive correlation with the patient’s age. About 15% of the variability in ferritin can be explained by the patient’s age. In effect, older patients had higher ferritin (r = 0.391, r2 = 0.153; P = 0.0025).
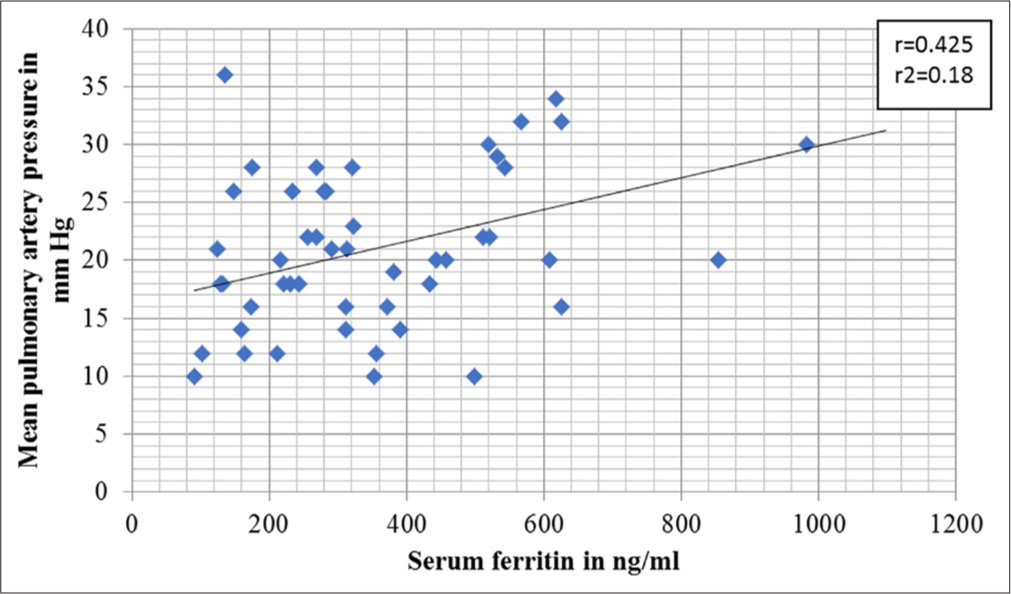
- Association of pulmonary hypertension and serum ferritin.
By multiple regression analysis, only 22% of the variability in MPAP levels could be predicted by Hb F, serum ferritin, number of transfusions, and Hb and HbA2/E levels (P = 0.02).
DISCUSSION
The median age of the study population was 21 years, and the majority did not receive any transfusion or had occasional transfusion till follow-up. We did not encounter short stature or growth retardation due to severe anemia, Hb below five g%, ferritin more than 2000 ng/mL, gall stones, leg ulcer, splenectomy, thrombosis, serious infections, unlike the severe form of Hb E beta-thalassemia.[7,8] In our study, those with high ferritin of more than 500 ng/mL showed a high incidence of pulmonary hypertension compared to those with <500 ng/mL ferritin. In the OPTIMAL CARE study, mean serum ferritin was 967.5 ± 853.9 ng/mL, and in Thailand, mean ferritin was 1,563.46 ng/mL, and 76% of patients had ferritin levels of more than 800 ng/mL.[9,10] Early age at presentation and low average Hb contributed to a high Mahidol score and a high incidence of pulmonary hypertension in our study. We detected pulmonary hypertension in 32% of NTD thalassemia cases who did not undergo splenectomy or any prior history of thromboembolic episodes. Selected patients who had high ferritin of more than 500 ng/mL were closely followed up every 3 months with echocardiography for the detection of new-onset pulmonary hypertension. In our cohort, low average Hb, early age of disease onset, obtaining first blood transfusion at a younger age, the requirement of frequent transfusion, splenomegaly, growth retardation, and high level of serum ferritin were more common in the moderate as compared to the mild group. In a study in Thailand, the E-SAAN score was used as a screening test for pulmonary hypertension risk in patients with NTDT.[11] There was good discrimination under the receiver operating characteristics curve of 0.88 with a 95% confidence interval of 0.8–0.95 in the validating group after using the cut-off point as a 4.5 score. As most of the study population was young, under 30 years old, and had no prior splenectomy, we could not estimate the E-SAAN score in the current study. The incidence of pulmonary hypertension in thalassemia intermedia was 24% in another study.[7] This was concordant with data in Lebanon, where pulmonary hypertension was a severe complication of 11–50%, leading to heart failure and death.[12,13] In our study, the participants who received more than three transfusion units had low Hb F, high Hb A2/E, and high ferritin. Advanced age and splenectomy as risk factors for pulmonary hypertension were widely accepted as described in the literature.[14] The majority of patients with high MPAP had early age at diagnosis, delayed initiation of blood transfusion, predominant male gender, low Hb,and high ferritin in our cohort. TRV of 2.5 m/s or higher on Doppler echocardiography was a strong predictor of death with a 40% mortality risk within 3 years of diagnosis.[15] We did not encounter right heart failure or sudden cardiac death even when serum ferritin was more than 1000 ng/mL, age above 40 years, and those with grade II pulmonary hypertension. All the patients were asymptomatic, and those with moderate pulmonary hypertension were started on either sildenafil or bosentan, along with calcium channel blockers. TRV was associated positively with age, cardiac volumes, and pulmonary regurgitation.[16] Even though none of the study participants underwent splenectomy, we detected asymptomatic pulmonary hypertension in 32%, similar to another study from Iraq where pulmonary hypertension was seen in 9% and not associated with splenectomy.[17,18] We found a positive association between increasing age and ferritin level as those who presented with ferritin more than 800 ng/ml were older adults compared to younger age patients with low serum ferritin, and this was one of the disease parameters that influenced the MPAP. In adult beta thalassemia, age and left atrial diameter were higher in splenectomized but showed lower ferritin levels.[19]
We identified compound heterozygosity for IVS 1-5(G>C)+CD26(G>A) as the most common beta gene mutation and αα/α3.7 as the most prevalent alpha gene deletion. Due to resource limitations, we did not evaluate for genetic polymorphisms or other disease-modifying gene mutations. Those who carried IVS1-5(G>C)+CD26(G>A) beta-globin mutation and alpha (-α)3.7 and -α4.2 deletion showed high Hb F in those who did not carry such beta gene and alpha gene mutations. Among patients with Hb E beta thalassemia, multiple single nucleotide polymorphisms associated with gamma-globin gene expression HBG2, BCL11A, HBS1L-MYB gene as well as those with KLF1 mutations exhibited mild phenotype compared to other rare genes such as COL4A3, DLK1, FAM186A, PZP, THPO, and TRIM51.[20,21] Homozygous or compound heterozygous beta thalassemia with the presence of concomitant alpha thalassemia has been shown to reduce the presence of excess alpha chains, thereby leading to a less severe phenotype. One example of this is the presence of XmnI polymorphism, one of three major Hb F quantitative trait loci, which increased gamma chain production and neutralized excess alpha chains, thereby causing Hb F variation and less severe phenotype. The presence of such XmnI polymorphism was not evaluated in the cohort due to study limitations.
In another published data on the genetic severity of E-beta thalassemia, heterozygosity for the presence of XmnI site polymorphism delayed disease onset and influenced the severity of phenotype.[22] Although the disease burden of Hb E beta thalassemia was high in the eastern pattern of India, due to resource constraints, barriers in access to centers specialized in thalassemia care, loss to follow-up of patients due to lack of support, and limited availability of genetic diagnosis, data on disease-modifying mutations in NTDT were scarce. Inheritance of β+ thalassemia mutations was associated with a less severe phenotype in another study.[23] Additional data on the association of beta-globin and alpha-globin deletion on various other complications and variable requirements of transfusion within the NTDT subset were lacking in the Indian population.
Median Hb F levels were lower in patients with morbidities in a study in NTDT.[24] In our study, high Hb F above 25% was seen in the majority with IVS 1-5 beta-globin gene mutation compared to other beta gene mutations and those with alpha globin gene deletions. In a large multicenter study, the risk of pulmonary hypertension in thalassemia intermedia is associated with splenectomy, previous episode of thromboembolic events, high nucleated red cell count, hydroxyurea or transfusion or chelation-naive status, but no significant difference was noted with coinheritance of alpha thalassemia or determinants associated with increased gamma chain production.[25] We detected low left ventricular ejection fraction in those who had pulmonary hypertension. In contrast, another study did not show any statistical differences in the number of transfusions, hematologic parameters, serum ferritin, and left ventricular ejection fraction in those who had pulmonary hypertension.[18] In a similar study, evaluating the effect of genotype on risk of pulmonary hypertension, genotype of thalassemia, splenectomy status, severity of anemia, and increasing age were risk factors, but serum ferritin, high nucleated red blood cell count, and age during first blood transfusion were not identified as risk factors. Similarly, the risk was more common in beta-thalassemia due to severe phenotype than in alpha-thalassemia or combined alpha and beta-thalassemia, as described in another study.[26] An increase in 1 g/dL of Hb was associated with a lower risk of complications such as leg ulcers, endocrinopathies, extra-medullary hematopoiesis, pulmonary hypertension, and more prolonged overall survival in a study.[27] The beneficial role of splenectomy to increase Hb was proven in earlier studies, but due to overwhelming sepsis and post-splenectomy complications, it is rarely preferred in resource-limited centers. We administered hydroxyurea at 10–20 mg/kg to all patients as Hb F induction by hydroxyurea has shown pronounced benefit in Hb E beta thalassemia, Hb Lepore, and XmnI polymorphism.[28] Luspatercept has demonstrated significant improvement in symptoms due to anemia when Hb is <10 g/dL in the BEYOND trial in NTDT,[29] and its potential disease-modifying benefits might change the outcome in South Asian countries.
Limitations
PCR for globin gene mutation was undertaken in only 50– 60% of the cases, and secondary and tertiary disease modifiers were not analyzed in our study. Despite the high burden of Hb E beta thalassemia, lack of right heart catheterization, cardiac/hepatic magnetic resonance imaging, ventilation-perfusion scan, and unavailability/resource constraints for novel biomarkers such as nitric oxide metabolites, prostaglandin, endothelin, soluble thrombomodulin, nontransferrin bound iron, N-terminal prohormone of brain natriuretic peptide, flow cytometry for circulating activated platelet and erythrocyte microparticles contributed to limited assessment of disease parameters and various complications except pulmonary hypertension.
CONCLUSION
Although young age, mild Mahidol score, high Hb F in a high proportion of beta mutation and alpha deletion, pulmonary hypertension was still a significant complication in its asymptomatic state even in the absence of splenectomy or prior thromboembolic episode in non-transfusion-dependent thalassemia. Furthermore, one-fourth of its variability is attributed to genotypic and hematologic parameters. Future multicenter and prospective clinical trials are warranted in resource-limited settings, emphasizing genetic modifiers on various complications in thalassemia as well as assisting in developing novel therapeutic agents.
Ethical approval
The study was approved by Institutional Ethics Commitee at Nil Ratan Sircar Medical College and Hospital, Kolkata, number NMC/000182 dated 7th January 2015.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480-7.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular genetics of beta-thalassemia: A narrative review. Medicine. 2021;100:e27522.
- [CrossRef] [PubMed] [Google Scholar]
- Modulatory effect of single nucleotide polymorphism in Xmn1, BCL11A and HBS1L-MYB loci on foetalhaemoglobin levels in beta-thalassemia major and Intermedia patients. J Pak Med Assoc. 2021;71:1394-8.
- [CrossRef] [PubMed] [Google Scholar]
- Non-transfusion-dependent thalassemia: A panoramic review. Medicina (Kaunas). 2022;58:1496.
- [CrossRef] [PubMed] [Google Scholar]
- Impact of genotype on multi-organ iron and complications in patients with non-transfusion-dependent ß-thalassemia intermedia. Ann Hematol. 2024;103:1887-96.
- [CrossRef] [PubMed] [Google Scholar]
- Link between genotype and multi-organ iron and complications in children with transfusion-dependent thalassemia. J Pers Med. 2022;12:400.
- [CrossRef] [PubMed] [Google Scholar]
- Leg ulcers: A report in patients with hemoglobin E beta thalassemia and review of the literature in severe beta thalassemia. Acta Haematol. 2022;145:334-43.
- [CrossRef] [PubMed] [Google Scholar]
- Survival and complications in patients with haemoglobin E thalassaemia in Sri Lanka: A prospective, longitudinal cohort study. Lancet Glob Health. 2022;10:e134-41.
- [CrossRef] [PubMed] [Google Scholar]
- Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The OPTIMAL CARE study. Blood. 2010;115:1886-92.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and risk factors for complications in patients with nontransfusion dependent alpha-and beta-thalassemia. Anemia. 2015;2015:793025.
- [CrossRef] [PubMed] [Google Scholar]
- A risk score for predicting pulmonary hypertension in patients with non-transfusion-dependent thalassemia in Northeastern Thailand: The E-SAAN score. Hematology. 2015;20:416-21.
- [CrossRef] [PubMed] [Google Scholar]
- A killer revealed: 10-year experience with beta-thalassemia intermedia. Hematology. 2014;19:196-8.
- [CrossRef] [PubMed] [Google Scholar]
- The natural history of thalassemia intermedia. Ann N Y Acad Sci. 2010;1202:214-20.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and risk factors for pulmonary arterial hypertension in a large group of ß-thalassemia patients using right heart catheterization: A Webthal study. Circulation. 2014;129:338-45.
- [CrossRef] [PubMed] [Google Scholar]
- A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011;365:44-53.
- [CrossRef] [PubMed] [Google Scholar]
- Echocardiographic evaluation of thalassemia intermedia patients in Duhok, Iraq. BMC Cardiovasc Disord. 2014;14:183.
- [CrossRef] [PubMed] [Google Scholar]
- Doppler-defined pulmonary hypertension in ß-thalassemia major in Kurdistan, Iraq. PLoS One. 2020;2020:e0243648.
- [CrossRef] [PubMed] [Google Scholar]
- Vascular and hemostatic alterations associated with pulmonary hypertension in ß-thalassemia hemoglobin E patients receiving regular transfusion and iron chelation. Thromb Res. 2019;174:104-12.
- [CrossRef] [PubMed] [Google Scholar]
- Cardiopulmonary testing in adult patients with ß-thalassemia major compared to healthy subjects. Ann Hematol. 2022;101:2445-52.
- [CrossRef] [PubMed] [Google Scholar]
- Whole exome sequencing and rare variant association study to identify genetic modifiers, KLF1 mutations, and a novel double mutation in Thai patients with hemoglobin E/beta-thalassemia. Hematology. 2023;28:2187155.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular analysis of non-transfusion dependent thalassemia associated with hemoglobin E-β-thalassemia disease without α-thalassemia. Mediterr J Hematol Infect Dis. 2019;11:2019038.
- [CrossRef] [PubMed] [Google Scholar]
- Hb E-ß-Thalassemia in Five Indian States. Hemoglobin. 2016;40:310-5.
- [CrossRef] [PubMed] [Google Scholar]
- Beta-thalassemia intermedia: A single thalassemia center experience from Northeastern Iraq [published correction appears in Biomed Res Int 2020;2020:2453270] Biomed Res Int. 2020;2020:2807120.
- [CrossRef] [PubMed] [Google Scholar]
- Fetalhemoglobin levels and morbidity in untransfused patients with ß-thalassemia intermedia. Blood. 2012;119:364-7.
- [CrossRef] [PubMed] [Google Scholar]
- Risk factors for pulmonary hypertension in patients with ß thalassemia intermedia. Eur J Intern Med. 2011;22:607-10.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of genotype on pulmonary hypertension risk in patients with thalassemia. Eur J Haematol. 2014;92:429-34.
- [CrossRef] [PubMed] [Google Scholar]
- Morbidity-free survival and hemoglobin level in non-transfusion-dependent ß-thalassemia: A 10-year cohort study. Ann Hematol. 2022;101:203-4.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical experience with fetalhemoglobin induction therapy in patients with ß-thalassemia. Blood. 2013;121:2199-372.
- [CrossRef] [PubMed] [Google Scholar]
- Luspatercept for the treatment of anaemia in non-transfusion-dependent β-thalassaemia (BEYOND): A phase 2, randomized, double-blind, multicentre, placebo-controlled trial. Lancet Haematol. 2022;9:e733-44.
- [Google Scholar]




